 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Estanis Navarro | -- | 4107 | 2022-06-20 10:41:44 | | | |
| 2 | Camila Xu | -22 word(s) | 4085 | 2022-06-29 03:19:23 | | | | |
| 3 | Camila Xu | + 22 word(s) | 4107 | 2022-09-13 04:01:15 | | | | |
| 4 | Camila Xu | -22 word(s) | 4085 | 2022-09-13 04:03:47 | | |
Video Upload Options
The 3′Untranslated regions (3′UTRs) of mRNAs, are non-coding regulatory platforms that control stability, fate and the correct spatiotemporal translation of mRNAs. Although initially considered as stabilizing features of the ORF, further work identified a number of new 3’UTR functions that controlled where, when and how mRNAs were translated. Furthermore, recent research has enriched the view of 3’UTRs from static regulators of mRNA translation to highly dynamic and modular regulatory platforms that respond to different stimuli by changing their structure. By using alternative polyadenylation and cleavage sites, alternative exons or by including exonized Alu cassettes, 3’UTRs modify their length, change their sequence and consequently their inventory of associated regulatory sites to establish different co-regulatory events.
1. Introduction
2. A Hint of History: Messenger RNAs (mRNAs) as Genetic Shuttles, from the Concept to the Molecule
In their seminal Nature paper of 1953 describing the double-helix structure of DNA, Watson and Crick had proposed that the genetic information of the organisms was encoded in the sequence of nuclear DNA [5], raising the question of how was this information transferred to the cytoplasm where protein synthesis took part. Later on, in 1958, Francis Crick hypothesized for a directional transfer of the genetic information inside the cell from DNA to RNA and to proteins, from then known as the "central dogma of molecular biology" [6]. Next, years of hectic work lead to the identification of a soluble and short-lived, unstable RNA fraction in E. coli cells infected with the bacteriophage T2r+ that showed a base composition similar to that of the phage DNA [7][8][9]. Although this RNA fraction was initially considered as a precursor of phage DNA, further work suggested that this could actually be the intermediate for the transmission of genetic information for the proteins that were synthesized during phage infection [8][9][10], a "messenger", or in the words of Jacob and Monod, "... a short-lived intermediate, or messenger, which becomes associated with the ribosomes where protein synthesis takes place [11]". This hypothesis was confirmed by Nirenberg and Matthaei after showing that addition of a synthetic polyribonucleotide template to a E. coli cell-free system stimulated aminoacid incorporation [12], a result that opened the door to the cracking of the genetic code. Lastly, work in a number of laboratories demonstrated that these “messenger” RNAs were not restricted to phage-infected E. coli cells, and similar RNA fractions were be also detected in eukaryotic cells from calf thymus [13], rat liver [14], mammary carcinoma cells [15] or HeLa cells [16].
On the other hand, isolation of the alpha/beta globin mRNAs led to the surprising finding that these were much longer than their coding potential required [17], and to the subsequent discovery that the mRNA fragments that actually encoded genetic information were flanked at their 5’ and 3’ ends by fragments of non-coding information that were heterogeneous in sequence and length [18][19], and included a 150-200 nt long poly-A tail segment at the end of the transcript [20][21][22] that was added post-transcriptionally [23][24]. This structure was shown to be shared by all eukaryotic mRNAs except those encoding histone proteins [25] (Figure 1 and see [26] for a historical account of the discovery of mRNA).

Figure 1. Structure of an eukaryotic mRNA and functions associated to the 3'UTR. AUG=initiation codon, RBP=RNA binding proteins, STOP=termination codons, PAS=polyadenylation signal, An=poly-A tail.
3. Sequences at the 3′UTR Regulate mRNA Stability and Function
The finding that 3′UTRs contained segments with a high degree of sequence-conservation across species [27] changed the view of mRNA-3′UTRs from simple stabilizing end-regions to complex, dynamic and highly structured regulatory platforms that responded to regulatory stimuli by generating sequence diversity. This allowed the expression of alternative regulatory boxes that controlled where, when and how mRNAs were translated, and established differential co-regulatory events. From a mechanistic point of view, the functional organization of mRNA-3′UTRs is based on short-sequence, cis-regulatory elements that are recognized and bound by trans-regulatory factors, either RNA binding proteins (RBPs) or other RNAs, mainly miRNAs [4]. Usually, 3′UTR-cis-acting sequences are structured in clusters of repeated modules that are recognized on a sequence basis by RBPs with repeated RNA-binding motifs (RBMs) or by miRNAs and other ncRNAs [4][28], although some of them are able to interact with the secondary structure of the binding motif [29].
Most messenger RNAs are targeted for selective and rapid degradation. The first motifs for mRNA stability described were the conserved AU-rich sequences (AREs) at the 3' UTR of unstable cytokine/chemokine mRNAs [30][31] (Figure 2). Basically, AREs are short sequences (50-150 bases long, see Figure 1) that include one to several copies of the pentanucleotide AUUUA in an AU-rich context, although a number of these destabilizing regions have been described that lack an AUUUA box [32]. The function of AUUUA-AREs is mediated by the binding of specific RNA Binding Proteins (RBPs). Other motifs have been associated with the control of mRNA stability. Thus a pyrimidine-rich sequence [(U/C)(C/U)CCCU] within the 3'UTR of tyrosine hydroxylase mRNA increased its stability during hypoxia [33], while on the contrary, a highly conserved UC-rich region (containing 5'-C(U)(n)C and 3'-CCCUCCC motifs) reduced stability of the androgen receptor (AR) by mediating binding of HuR and Poly(C)-binding proteins-1 and -2 (CP1 and CP2) to the 3-UTR of the AR mRNA [34].
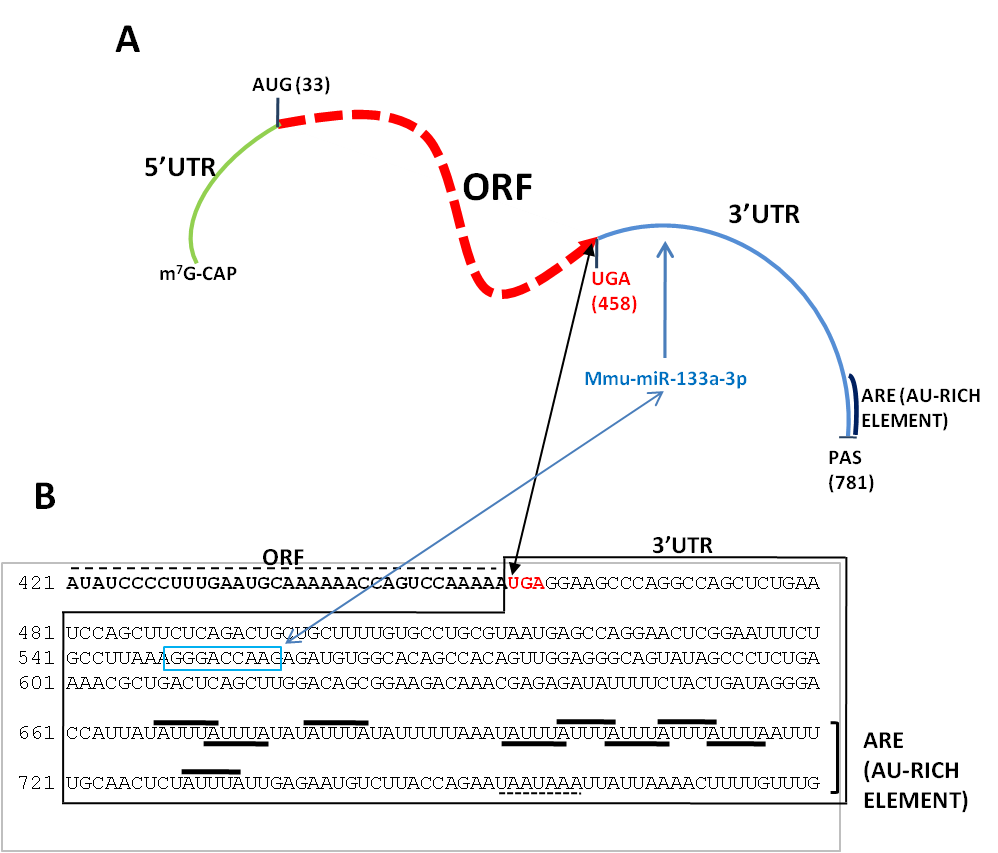
Figure 2. Structure of the murine mRNA for granulocyte-macrophage colony stimulating factor (GM-CSF). (A) Diagram showing different structural elements of the mRNA: 5'/3'UTR=5'/3' untranslated regions, ORF=open reading frame, PAS=polyadenylation signal. Shown are also the initiation (AUG) codon at position 33 and the STOP (UGA) codon at position 458. Not drawn to scale. (B) Sequence of the 3'UTR of the murine GM-CSF mRNA (Genbank X03019) showing different stability determinats, a mmu-miR-133a-3p binding sequence (boxed in light blue) and an AU-rich element composed by several AU motifs (black bars). The polyadenylation signal (AAUAAA) is underlined. As modified from [35].
3.1. Stability Determinants in the 3′UTRs of Eukaryotic mRNAs Recognized by RNA Binding Proteins or microRNAs
The intracellular fate of mRNAs depends on the complex interplay among degradation or stabilization-promoting RBPs, whose equilibrium determines mRNA half-life, as in the case of the vascular endothelial growth factor mRNA whose stability in response to hypoxia relied on the balance among degradation-promoting ARE-binding factors, such as the ACTH-regulated zinc-finger proteins Tis11,Tis11b and Tis11d [36], and the stabilization factors HuR that bound to U-rich motifs [37] or hnRNPL that interacted with an AC-rich sequence [38], all of them in the 3’UTR. Other RBPs have been described to affect mRNA stability by binding to sequential motifs at their 3’UTRs, such as members of the CCCH zinc finger [39], hnRNP [40] or ELAV [41] families (see [42] for a more detailed description).
3.2. Spatial-Temporal Regulation of mRNA Function: Maternal mRNAs in the Maternal-to-Zygotic Transition
Control of mRNA function by RBPs and miRNAs is a very dynamic process that includes temporal or spatial regulatory axes. A relevant model to study the mechanisms of spatial-temporal regulation of mRNA translation is the oocytic cell, whose first stages of development are regulated in the absence of transcription by a pool of mRNAs (maternal mRNAs) that are loaded onto the cell cytoplasm and account for over 7000 different transcripts [53]. A subset of these maternal mRNAs is maintained functionally inactive in oocytes through the binding of RBPs to specific 3′UTR motifs and AU-rich cytoplasmic polyadenylation elements (CPEs) [54]. In the maternal-to-zygotic transition (MZT) these are then submitted to cytoplasmic polyadenylation to activate translation of mRNAs encoding transcriptional regulators, among others [55]. Once translational repression is released, transcription is activated and maternal mRNAs are degraded by the deadenylation machinery [56][57] and the poly-A specific ribonuclease subunit 2 (PAN2) [58]. Deadenylation results in mRNA destabilization, translational inactivation [59] and the clearance of maternal mRNAs by the poly-A-specific exoribonuclease (PARN) [60], miR-430 [61] and the RNA m6A-reader, YTHN6-methyladenosine RNA binding protein 2 (YTHDF2) [62].
On the other hand, appropriate spatial translation is ensured by restricting the intracellular distribution of mRNAs through the binding of specific RBPs. Genetic studies in a number of cell systems have provided some insights on the mechanisms and genes promoting and maintaining an asymmetric subcellular distribution of certain mRNAs (see [63] for a recent review). In this sense, the body plan of Drosophila can be traced down to the first stages of oogenesis, in which a complex interplay of regulatory mechanisms in the form of RNPs produced by the neighbour nurse cells, ensure the loading and asymmetric intracellular distribution of maternal determinants, their precise translational regulation, and the asymmetric cell divisions needed to generate daughter cells with distinct cytoplasmic contents and developmental fates [64]. Briefly, the establishment of an antero/posterior (a/p) axis is controlled by the asymmetric loading of the bicoid (bcd) mRNA to the anterior pole, and of nanos (nos) and oskar (osk) mRNAs to the posterior pole of the oocyte in a way dependent on the microtubule network, on molecular motors of the dynein and kinesin families [65], as well as on the presence of a conserved YUGUUYCUG box in the 3′UTRs of bicoid and nanos mRNAs [66]. Fertilization will activate translation of bicoid and nanos so that the asymmetric distribution of their protein products will generate a functional a/p axis.
4. Regulation of 3’UTR Length by Alternative Polyadenylation or Alternative Splicing
The sequencing revolution has evidenced the high degree of variation in the length of 3'UTRs of mRNAs, and its subsequent impact on the regulatory landscapes of mRNA function. Regulation of 3’UTR length is thus becoming an important research topic in the control of gene expression by its potential to regulate mRNA-protein or mRNA–RNA interactions, and consequently mRNA function [67]. In this section researchers will deepen on the mechanisms known to regulate 3'UTR length by alternative polyadenylation and cleavage (APA) or alternative splicing (AS).
4.1. Regulating 3’UTR Length by Alternative Polyadenylation
Pre-messenger RNAs (pre-mRNAs) are specifically cleaved and polyadenylated at precise positions of their 3’ ends in a way determined by a specific polyadenylation signal (PAS) and executed by a number of multiprotein complexes. The polyadenylation signal (AAUAAA in its canonical form) positions the cleavage/polyadenylation specificity factor complex (CPSF) close to the cleavage site, over 30 nts from the specific site at which the pre-mRNA will be cleaved and the poly(A) tail will be added by the PAP activity ([68] for a review). Cleavage precision is ensured by the presence of two U-rich, upstream (USE) and downstream (DSE) sequence elements next to the AAUAAA signal that help to distinguish functional polyadenylation sites from randomly occurring hexamers [69]. These U-rich sites are recognized by the multiprotein complexes cleavage factor I (CFI) and cleavage/polyadenylation specificity factor (CPSF) which bind to the U-rich USE and AAUAAA hexamer, respectively, and the cleavage stimulation factor CstF binding to the U-rich DSE [70]. Over 70% of human genes have more than one polyadenylation site in their 3'UTRs and 50% have three or more [71], while in mouse liver over 60% of expressed genes harbour multiple polyadenylation signals in their 3'UTRs [72]. Use of different sites makes alternative polyadenylation a widespread mechanism to regulate gene expression by generating transcript variants that are heterogeneous in length, show alternative 3' ends [73] and differential functional features as the microRNA binding potential [74] among others (Figure 3). The emerging picture on the use of alternative PASs proposes that 3’UTR shortening would increase mRNA stability by relaxing protein or miRNA-based mechanisms of mRNA degradation, while 3’UTR lengthening would strengthen accessibility to miRNAs [75]. While long 3’UTRs have been mostly detected in quiescent stem cells, differentiated cells, or early in development, shortened 3UTRs have been mostly described in quickly cycling cells such as proliferative stem/progenitor cells or tumour cells (see [42] for a recent review].
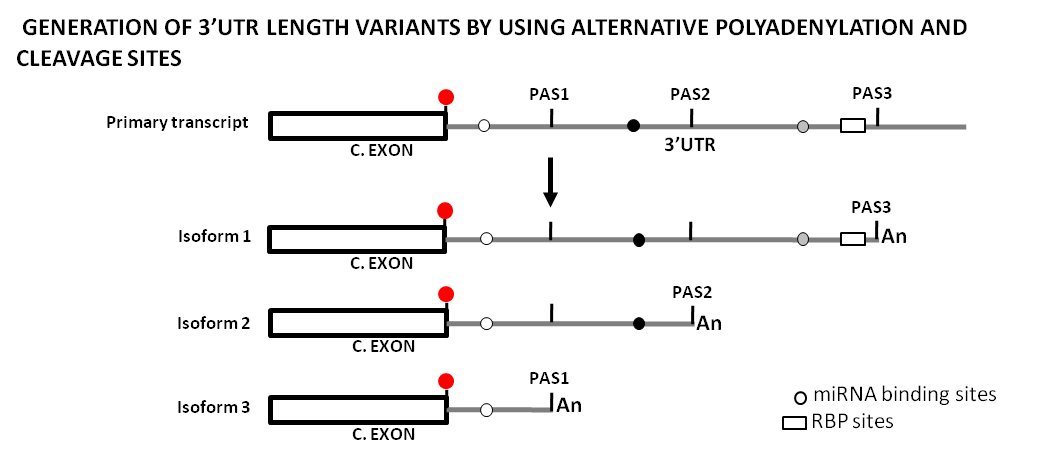
Figure 3. Original drawing showing the use of alternative polyadenylation and cleavage sites to generate sequence length variability at 3’UTRs and its potential impact on the regulatory landscape by miRNAs and RBPs. This diagram represents the generation of three different transcript isoforms from a primary transcript by the use of three different polyadenylation and cleavage sites (PAS1, PAS2, PAS3), and how this changes the mRNA regulatory landscape. Shown is a terminal coding exon with its stop codon (red dot), as well as the 3'UTRs (gray lines) with the different PAS, miRNA binding sites (gray dots), and a RBP site (white box). [An] stands for the poly-A tail. C.EXON stands for Coding Exon.
APA-depending shortening of 3’UTRs is caused by changes in the expression of different components of the cleavage/polyadenylation complexes, i.e., cleavage factors CFI (composed by CPSF5/CFIm25/NUDT21, CPSF6-7) and CFII (PCF11, CLP1), cleavage and polyadenylation specificity factors CPSF (CPSF1-4, WDR33, FIP1L1) or cleavage stimulation factors CSTF1-3 ([76] for review). In this sense, Masamha et al. characterized CPSF5/CFIm25/NUDT21 as an inhibitor of proximal polyadenylation [77], and down-regulation of CPSF5/CFIm25/ NUDT21 was seen to increase transcript stability in lung adenocarcinomas and lung squamous cell carcinomas by promoting shortening of the 3’UTRs of IGF1R, CCND1 and GSK3β mRNAs [78] while other factors such as Pcf11 and Fip1 [79], CPSF6 [80], CSTF2 and CPEB3 [81], or HuD also favored the usage of proximal polyadenylation signals to generate shorter 3’UTRs [82]. On the contrary, other factors such as SRSF3 directly promoted the use of distal PAS [83].
4.2. Regulating 3’UTR Length by Alternative Splicing
Splicing-based mechanisms, such as alternative or cryptic splicing of terminal untranslated exons or the integration of repetitive elements through exonization also have the potential to generate 3’UTR length variants ([84][85] and Figure 4). Nevertheless, the impact of splicing-based mechanisms of 3’UTR lengthening is lower than those based in alternative polyadenylation and cleavage, as highlighted by a bioinformatic analysis on the superfamily of odorant receptor (OR) genes that showed that over 80% of OR mRNAs were submitted to alternative polyadenylation while only a few of these used alternative splicing to generate variant 3’UTRs [86]. Regulation of alternative splicing is a very complex topic that involves multiple regulatory sites at the pre-mRNAs (splicing donor and acceptor sites, canonical, cryptic and alternative sites, splicing enhancers and silencers, etc.) that are recognized by a plethora of mRNA binding proteins, U-small nuclear RNAs and associated proteins ([87] for review). In this sense, and as a general rule, introns would be identified by the binding of U1snRNP and the U2AF65/U2AF35 complex to the splice sites [88]. Basically, a few splicing mechanisms originate 3’UTR length variability and modify 3 ‘UTR regulatory potential, i.e., intron retention, exon skipping, incorporation of one of two mutually exclusive terminal exons of different length or activation of cryptic splice sites that modify the relative lengths of the ORF and 3’UTR ([89] and Figure 4). In this sense, a number of splicing regulators have been implicated in the regulation of 3’UTR length. Thus, cytoplasmic polyadenylation element binding protein1 (CPEB1) was seen to mediate shortening of 3’UTRs by changing patterns of alternative splicing through repression of U2AF65 recruitment or by influencing the use of alternative polyadenylation sites [90], and splicing of the 3’UTR of Yes1-associated transcriptional regulator (YAP) mRNA was seen to be dependent on hnRNPF [91]. Furthermore, SR protein kinase SPK-1 promoted 3’UTR splicing of polarity protein Par-5 mRNA [92], and quaking (QK), a global regulator of splicing [93], was seen to promote stability of hnRNPA1, a repressor of alternative splicing, by binding a conserved 3’UTR sequence [94]. Lastly, expression of splicing factors ESRP1, PTB and SF2/ASF, was significantly altered in cardiac hypertrophy, leading to removal of instability-promoting AT-rich elements from 3’UTRs [95]. As for the case of APA, these splicing-based changes of 3’UTR length were associated with changes in the binding patterns of miRNAs. A special case of 3’UTR-lengthening caused by alternative splicing is the retention of non-coding introns [96]. Researchers reported that a significant number of transcripts included retained introns in their 3’UTRs that harbored miRNA binding sites (in 387 out of the 2864 human genes analyzed [97]), or Staufen2 (Stau2) sites (in 356 transcripts [98]). Interestingly the presence of an alternative retained intron in the 3’UTR of splicing factor SRSF1 (ASF/SF2) protected this isoform from NMD degradation in HCT116 colon cancer cells [99].
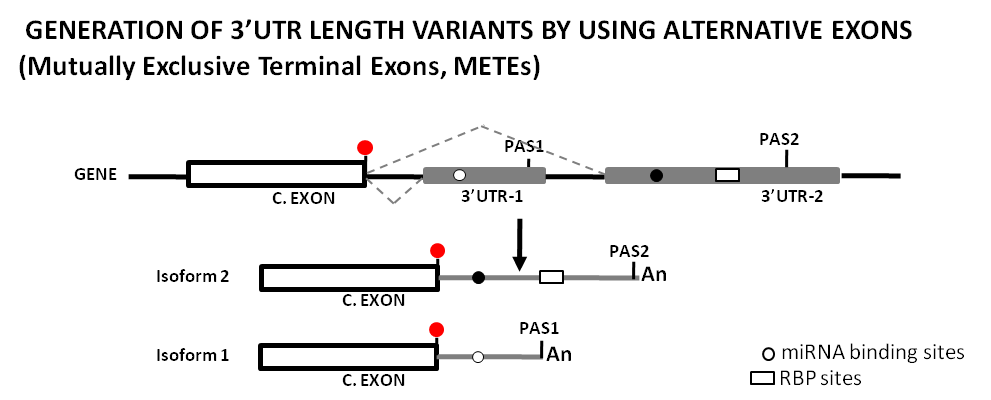
Figure 4. Original drawing showing the use of of alternative splicing sites (as Mutually Exclusive Terminal Exons, METEs) to generate sequence length variability at 3’UTRs and its potential impact on the regulatory landscape by miRNAs and RBPs. This diagram represents the generation of two different transcript isoforms from a single gene by the use of two mutually exclusive 3'UTR terminal exons, and how this changes the mRNA regulatory landscape. Shown is a terminal coding exon with its stop codon (red dot), as well as the 3'UTRs (gray boxes/lines) with the different PAS, miRNA binding sites (gray dots), and a RBP site (white box). Dotted gray lines between exons show the two splice events produced. [An] stands for the poly-A tail. C.EXON stands for Coding Exon.
5. Regulation of 3’UTR Length by Exonization of Repeated Sequences of the Alu Family
Mammalian 3’UTRs harbor a number of mobile genetic elements from the Short/Long Interspersed Nuclear Element (SINE/LINE) families, and among them the family of Alu repeated sequences [100]. Alu elements can be found in clusters in intergenic or intronic regions, or embedded in transcriptional units, mostly in the 3’UTRs of mRNAs but also in their 5’UTR or coding regions [101]. Although most of the Alu elements are currently stable genomic fossils, a very small number of Alu elements (called “young” or “master”Alus) are still retrotransposition-competent [102]. Alu repeats can impact on mRNA function by different mechanisms, and among them by “exonization”, by which the splicing machinery incorporates, “de novo”, an intronic Alu element to a mature transcript with the subsequent structural modifications derived of the functional activation of alternative stop-codons and polyadenylation signals encoded in or downstream of the Alu element ([103] and Figure 5).
5.1. Docked-Alu Elements as miRNA Targets
3’UTR-Alu repeats have been proposed as potential sites for specific miRNA binding [104] with target sites coinciding with conserved Alu sequences [105], although this is a highly controversial topic and other researchers consider these Alu-dependent miRNA binding sites as neutral or non-functional [106]. In this sense, only a few 3’UTR-Alu/miRNA interactions have been confirmed, among them the targeting of double minute 2/4 mRNAs (Mdm2/4) by miR-661 [107], or that of RAD1, GTSE1, NR2C1, FKBP9 and UBE2l by miR-15a-3p and miR302d-3p [108]. Other researchers consider that these 3’UTR-Alus could actually work as miRNA sponges [109] in a way similar to that proposed for free Alus [110] or other ncRNAs [111]. In addition to the sequence-specific determinants of mRNA stability above described, other sequence-independent mechanisms have been reported that rely on the generation of Alu-dependent secondary structures in the 3’UTRs and are recognized by the A-to-I edition machinery [112], or by the Staufen-mediated Decay (SMD) pathway of mRNA degradation [113].
5.2. Impact of Exonized Alus on the 3’UTRome: Generating Alternative Stop Codons, Polyadenylation Signals or Splicing Sites
Alu repeats modify the 3’UTRome through the double process of exonization and subsequent neo-functionalization of the exonized sequence [114]. Alu-repeated elements contain a number of potential 5’/3’ splice sites that facilitate their incorporation to 3’UTRs by alternative splicing [115], although these are suboptimal variants of the canonical splicing donor/acceptor signals and suggest that expression of the Alu-including variants would not be constitutive, but optional [116]. The process of Alu-exonization is relatively frequent and some researchers have estimated that over 5% of alternative exons in the human genome could derive from Alu elements [117], with over 300 of these (corresponding to 243 genes) leading to the formation of new 30 terminal variants [118]. At the mechanistic level, exonized Alus generate gene variants with alternative 3’ ends through the activation of premature stop codons [119] or through the activation of downstream cryptic polyadenylation sites (PAS) [120] that can truncate or elongate a mature transcript depending on the location of the Alu-derived sequence (Figure 5).

Figure 5. Original drawing showing the exonization of an Alu cassette to generate sequence length variability at 3’UTRs and its potential impact on the regulatory landscape by miRNAs and RBPs. This diagram represents the generation of two different transcript isoforms from a single gene by the exonization of an Alu repeated element and how this changes the mRNA regulatory landscape. The Alu cassette harbors cryptic splice sites (CSSs) originated by mutation or activated by the unbalance of splicing regulatory elements (see main text for details). In this diagram, exonization of the Alu cassette lead to a change in the ORF and to the activation of a premature stop codon and PAS (shown as *). Shown is the 3’ end of an ideal gene and the two transcripts generated from it. The stop codon is shown as a red dot, the 3'UTRs as gray boxes/lines with the different PAS, miRNA binding sites (gray dots), and a RBP site (white box). Dotted gray lines between exons show the two splice events produced. [An] stands for the poly-A tail. C.EXON stands for Coding Exon.
Over 10,000 Alu elements are harbored in 3’UTRs of human protein-coding genes, of which more than one hundred have the ability to reprogram the 3’UTR length by providing functional polyadenylation and cleavage sites (PAS) to their transcripts [121]. These 3’UTR-Alus are mainly found in the forward sense orientation and show hot-spots of PAS-cleavage signals [118], mainly in their A-rich linker region between the two Alu arms, or in the short polyA tails of the Alu elements which can mutate to canonical AAUAAA polyadenylation signals [122]. The molecular mechanisms promoting the inclusion of exonized Alus into mature transcripts are complex and poorly studied [123]. A first mechanism relies in the competition for cryptic and functional splice sites between the exonization-promoting splicing factor U2AF65 and the suppressive hnRNP-C1/C2 that displaces the former from splice sites and represses exonization. Deregulation of this process, e.g., by mutations in hnRNP-C binding sites, would cause the aberrant activation of cryptic splice sites and result in Alu exonization [124]. Lastly, Alu exonization has been shown to be altered in a number of human diseases (see [42] for a recent review on this topic).

