 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Subir Sarker | + 3636 word(s) | 3636 | 2020-10-04 18:25:37 | | | |
| 2 | Rita Xu | -869 word(s) | 2767 | 2020-10-12 08:33:24 | | |
Video Upload Options
Wild animals harbour a large number of adenoviruses that remain uncharacterised with respect to their genomic organisation, diversity, and evolution within complex ecosystems. Here, we discovered the first complete genome sequence of an atadenovirus from a passerine bird that is tentatively named Passerine adenovirus 1 (PaAdV-1). The PaAdV-1 genome is 39,664 bp in length, which was the longest atadenovirus to be sequenced, to the best of our knowledge, and contained 42 putative genes. Its genome organisation was characteristic of the members of genus Atadenovirus; however, the novel PaAdV-1 genome was highly divergent and showed the highest sequence similarity with psittacine adenovirus-3 (55.58%). Importantly, PaAdV-1 complete genome was deemed to contain 17 predicted novel genes that were not present in any other adenoviruses sequenced to date, with several of these predicted novel genes encoding proteins that harbour transmembrane helices. Subsequent analysis of the novel PaAdV-1 genome positioned phylogenetically to a distinct sub-clade with all others sequenced atadenoviruses and did not show any obvious close evolutionary relationship. In contrast to the previous studies where authors proposed the reptilian origin of atadenoviruses, our resulting tress consistently demonstrated that atadenoviruses might have first evolved in a bird species that was present before the passerines and psittacine clades separated and were the ancestor of both clades. Further investigations on the structure and function of its major proteins and extended studies on closely related species can be suggested to broaden the knowledge in host specificity for adenovirus infection.
1. Introduction
Adenoviruses are medium-sized, non-enveloped, linear, double-stranded DNA (dsDNA) viruses within the family Adenoviridae [1]. The family Adenoviridae contains five accepted genera [1]. One of these genera, the Atadenovirus, was added in 2002, to include adenoviruses that were previously assigned to the genus Mastadenovirus, but varied significantly based on genomic size, structure, genes, and gene arrangement. Originally all the viruses in the genus were thought to have an A+T content bias [2][3], but with the discovery of new species in this genus, it has been shown that the A+T bias is not a consistent feature of this genus [4][5][6]. The size of sequenced atadenovirus genomes ranges between 27 and 34 kb, and all have the characteristic inverted terminal repeat (ITR) found in all adenoviruses [7]. Atadenoviruses have a set of core genes shared with the other adenovirus genera plus genus-specific genes. A feature of adenoviruses is their ability to acquire genes from their hosts, bacteria, fungi, and other viruses. The Atadenoviruses appear to be particularly adept at this and all atadenoviruses sequenced to date contain five or more genes acquired from other organisms or whose origin is not known [5][8][9][10]. These genes are diverse in their function and appear to be lost as often as they are acquired as the atadenoviruses evolve.
Atadenoviruses have been detected in a diverse range of hosts, including birds [4][11][12][13][14][15][16][17][18], reptiles (order Squamata; lizards, snakes, and worm lizards), ruminants [8][9][15][19], marsupials [20][21], and a common tortoise [22]. Using a partial DNA polymerase gene sequence, recent studies also report the presence of a large number of novel atadenoviruses circulating in wild passerine species of birds in Australia and Europe and passerine species kept in aviculture collections [4][17][18]. Because of the limited sequence information for these passerine adenoviruses, there is still considerable uncertainty about their phylogenetic relationship to each other and other atadenoviruses. Additionally, only two avian atadenoviruses, the psittacine adenovirus 3 (PsAdV-3) and duck atadenovirus (DAdV), have been fully sequenced to date [11][23][24].
2. Genome of PaAdV-1
The assembled passerine adenovirus 1 (PaAdV-1) complete genome was a linear double-stranded DNA molecule of 39,664 bp in length, which was the longest atadenovirus to be sequenced, to the best of our knowledge. Like most atadenoviruses, the PaAdV-1 genome contained a central conserved coding region bounded by two identical inverted terminal repeat (ITR) regions. The length of the ITR varies considerably in other atadenoviruses and ranges from 40 to 194 bp long [9][10]. The ITR of PaAdV-1 encompassed 193 bp each with the coordinates of 1–193 sense orientation and 39,450–39,642 antisense orientation. The novel PaAdV-1 genome sequence was shown to contain a balanced G+C percentage (53.70%). The known AdV genomes that were most closely related to the PaAdV-1, according to complete genome analysis, were psittacine adenovirus 3 (PsAdV-3; 55.58%), snake adenovirus 1 (SnAdV-1; 55.58%), bearded dragon adenovirus 1 (BDAdV-1; 55.20%), and duck adenovirus A (DAdV-A; 54.25%).
3. Genome Annotation and Comparative Analyses of PaAdV-1
The PaAdV-1 genome had 42 predicted methionine-initiated ORFs encoding proteins that were annotated as putative genes and were numbered from left to right (Figure 1 and Table 1). Comparative analysis of the protein sequences encoded by the predicted ORFs, using BLASTX and BLASTP, identified homologs with significant protein sequence similarity (E value ≤ 10−4) for 25 ORFs (Table 1), while 17 ORFs (ORF01-04, ORF6-18) were found to be unique according to the BLAST database. Among the predicted protein-coding ORFs of the PaAdV-1 genome, 24 were homologs to other AdVs gene products (Table 1). Among these homologues AdVs gene products, the highest number of protein-coding genes (21) in PaAdV-1 demonstrated homologs to the psittacine adenovirus-3 (PsAdV-3). The remaining three genes, encoding E1B-large T-antigen, E4, and a hypothetical protein (RH0, F-box related protein), were homologues to amniota adenovirus 1 (protein identity 25.26%, GenBank accession no. QEJ80749.1), DAdV-1 (protein identity 26.92%, GenBank accession no. AJA72340.1/), and BAdV-D (protein identity 30.15% GenBank accession no. NP_899151.1/), respectively. The gene product of ORF05 was predicted to encode a 92 amino acid (aa) (molecular weight/theoretical isoelectric point, Mw/pI-10.27 kDa/11.16). This protein was predicted to contain a protein homolog to prokaryotic ankyrin repeat domain-containing protein-50 by BLAST search with a >49% (query coverage 61% and E-value: 2.00 × 10−5) amino acid sequence similarity.
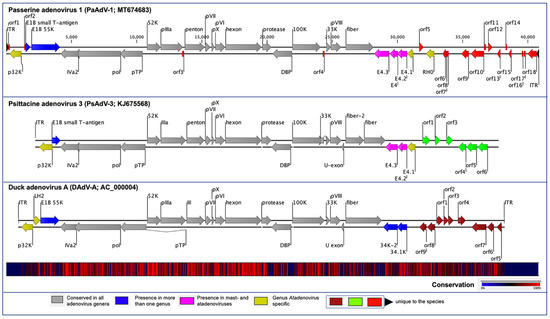
Figure 1. Schematic illustration of the avian atadenoviruses. Schematic map of the passerine adenovirus 1 (PaAdV-1, GenBank accession no. MT674683), in comparison with duck adenovirus A (DAdV-A, GenBank accession no. AC_000004) and psittacine adenovirus 3 (PsAdV-3, GenBank accession no. KJ675568), using CLC Genomic Workbench (version 9.5.4, CLC bio, a QIAGEN Company, Prismet, Aarhus C, Denmark). The arrows symbolize adenovirus genes and open reading frames (ORFs) predicted to code for proteins, indicating their direction of transcription. Each gene or ORF is colour coded, as indicated by the colour key in the legend. The bottom graph represents the sequence conservation between the aligned PaAdV-1, DAdV-A, and PsAdV-3 sequences at a given coordinate at each position in the alignment. The gradient of the colour reflects the conservation of that particular position is in the alignment. Red presents 100% conservation across all three viruses, black 50% conserved regions, and blue less than 50% conserved regions.
Table 1. Predicted protein-coding genes of PaAdV-1.
| PaAdV-1 Synteny | Start (nt) | Stop (nt) | Strand | Size (aa) | PsAdV-3 Synteny |
|---|---|---|---|---|---|
| ORF01 | 150 | 263 | + | 37 | |
| p32K | 250 | 1161 | − | 303 | p32K |
| ORF02 | 1368 | 1469 | + | 33 | |
| E1B protein, small T-antigen | 1466 | 1849 | + | 127 | E1B protein, small T-antigen |
| E1B protein, large T-antigen | 1911 | 4049 | + | 712 | |
| IVa2 protein | 4094 | 5482 | − | 462 | IVa2 protein |
| DNA polymerase | 5164 | 8634 | − | 1156 | DNA polymerase |
| pTP | 8568 | 10,208 | − | 546 | pTP |
| 52K protein | 10,547 | 11,581 | + | 344 | 52K protein |
| pIIIa protein | 11,574 | 13,259 | + | 561 | IIIa protein |
| ORF03 | 13,201 | 13,350 | − | 49 | |
| penton protein | 13,405 | 14,916 | + | 503 | penton protein |
| pVII protein | 14,942 | 15,289 | + | 115 | pVII protein |
| pX protein | 15,384 | 15,587 | + | 67 | pX protein |
| pVI protein | 15,651 | 16,403 | + | 250 | pVI protein |
| hexon protein | 16,421 | 19,150 | + | 909 | hexon protein |
| Protease | 19,221 | 19,901 | + | 226 | protease |
| DBP | 19,999 | 21,486 | − | 495 | DNA-binding protein |
| 100K protein | 21,508 | 23,607 | + | 699 | 100K protein |
| ORF04 | 23,769 | 23,897 | − | 42 | |
| 33K protein | 24,044 | 24,391 | + | 115 | 33K protein |
| pVIII protein | 24,379 | 25,053 | + | 224 | pVIII protein |
| fibre protein | 25,407 | 27,509 | + | 700 | fibre 2 protein |
| E4.3 protein | 27,555 | 28,685 | − | 376 | E4.3 protein |
| E4 protein | 28,737 | 29,360 | − | 207 | |
| E4.2 protein | 29,299 | 30,036 | − | 245 | E4.2 protein |
| E4.1 protein | 30,058 | 30,513 | − | 151 | E4.1 protein |
| ORF05 | 30,907 | 31,185 | + | 92 | |
| RH0 | 31,341 | 31,958 | − | 205 | |
| ORF06 | 32,572 | 32,808 | − | 78 | |
| ORF07 | 32,762 | 32,902 | − | 46 | |
| ORF08 | 32,896 | 33,015 | − | 39 | |
| ORF09 | 33,901 | 34,446 | − | 181 | |
| ORF10 | 34,604 | 35,542 | − | 312 | |
| ORF11 | 35,541 | 35,672 | + | 43 | |
| ORF12 | 35,828 | 36,118 | + | 96 | |
| ORF13 | 36,429 | 36,683 | − | 84 | |
| ORF14 | 37,082 | 37,195 | + | 37 | |
| ORF15 | 37,306 | 37,527 | − | 73 | |
| ORF16 | 38,325 | 38,432 | − | 35 | |
| ORF17 | 38,483 | 38,611 | − | 42 | |
| ORF18 | 38,767 | 39,480 | − | 237 |
The orientation of the predicted conserved genes in the PaAdV-1 was identical to that of PsAdV-3 and DAdV-A (Figure 1). The left-hand (LH) region of the PaAdV-1 genome contained a genus Atadenovirus specific gene homologue of p32K (Figure 1), followed by genes encoding E1B small T-antigen and E1B large T-antigen that were homologues to PsAdV-3 and DAdV-A, respectively. The amino acid sequence similarity of p32K was relatively low, as compared to other atadenoviruses, ranging from 30.83% to 33.51%, where the highest similarity was demonstrated with SnAdV-1 (33.51). Open reading frames corresponding to conserved LH proteins present in other atadenoviruses were not found in PaAdV-1.
At the centre of the PaAdV-1 genome, all the expected AdVs conserved genes were found, and their degree of homology with other atadenoviruses is shown in Figure 1 and Table 1. The only expected gene that was not found was the U-exon gene. The maximum similarity of individual proteins of PaAdV-1 to homologs in other atadenoviruses varied significantly and was not predictable (Table 2). For example, the DNA polymerase and penton base protein showed the highest pairwise identity with homologous proteins from PsAdV-3, whereas hexon, DNA binding protein, and fibre protein displayed the highest match with DAdV-A. Among the major capsid proteins, fibre protein showed a low amino acid identity, ranging from 15.65% to 25.42%, whereas penton base and hexon exhibited a high amino acid identity with PsAdV-3 (75.94%) and DAdV-A (80.04%), respectively. Furthermore, the additional predicted conserved proteins analysed also demonstrated high identity with homologous atadenoviruses proteins from other species (Table 2).
Table 2. Comparative G+C (%) content and pairwise identity of representative atadenovirus species against passerine adenovirus 1 (PaAdV-1) on the basis of complete genome nucleotide sequences and selected core proteins amino acid sequences. Shading and bold front highlights maximum similarity. Selected proteins were aligned by using MAFFT in Geneious (version 10.2.2, Biomatters, Ltd., Auckland, New Zealand) with the chosen of following parameters to calculate % similarity: scoring matrix BLOSUM62, Gap open penalty = 1.53. Blosum62 with threshold 1 (percentage of residues which have score > =1 in the Blosum62 matrix).
| Reference Atadenoviruses | Genome Identity (%) | G+C Content | % Pairwise AA Similarity with PaAdV-1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| p32K | IVa2 | DNA Pol | pTP | Penton | Hexon | Protease | DBP | Fibre Protein | ||||
| Avian | PaAdV-1 | 53.7 | ||||||||||
| PsAdV-3 | 55.58 | 53.5 | 30.83 | 59.01 | 53.45 | 33.28 | 75.94 | 76.44 | 56.19 | 36.02 | 17.52 | |
| DAdV-A | 54.25 | 45.5 | 32.78 | 64.56 | 52.89 | 32.32 | 73.57 | 80.04 | 53.1 | 36.97 | 25.42 | |
| Ruminant | BAdV-D | 53.38 | 37.1 | 29.73 | 70.35 | 52.73 | 32.37 | 72.69 | 76.09 | 56.19 | 35.77 | 22.38 |
| OAdV-7 | 53.69 | 33.6 | 33.53 | 69.97 | 51.71 | 32.77 | 73.29 | 77.89 | 53.98 | 36.07 | 23.27 | |
| OdAdV-1 | 53.27 | 33.3 | 32.65 | 63.59 | 52.09 | 31.45 | 73.35 | 77.32 | 54.42 | 36.08 | 23.99 | |
| Reptilian | LAdV-2 | 53.43 | 47.8 | 32.47 | 59.23 | 52.39 | 33.38 | 72.91 | 76.64 | 56.64 | 35.45 | 15.65 |
| SnAdV-1 | 55.58 | 56.8 | 33.51 | 57.94 | 50.34 | 32.49 | 72.63 | 75.33 | 58.41 | 34.5 | 20.06 | |
| BDAdV-1 | 55.2 | 56.6 | 33.15 | 58.80 | 50.70 | 33.14 | 69.18 | 75.84 | 58.08 | 31.72 | 24.35 | |
In the right-hand (RH) region of PaAdV-1, ORFs corresponding to four E4 genes were found (Figure 1 and Table 1). Among them, three (E4.1, E4.2, and E4.3) exhibited the greatest amino acid homology with proteins from PsAdV-3 (protein identity ranging between 27% and 35%), whereas one (E4) had the greatest amino acid homology with proteins from DAdV-1 (26.92% amino acid identity). To the right of the E4 region, the PaAdV-1 genome contained only one ORF previously described in atadenoviruses. This ORF codes for a protein of 205 residues that appears to be a homolog of the F-box domain found in BAdV-D (protein identity 30.15%). The RH0 gene was followed by 13 ORFs whose predicted protein products have not been identified in other adenoviruses previously (Figure 1).
4. Unique ORFs in PaAdV-1
The PaAdV-1 genome encoded all the conserved genes present in other adenoviruses, except the U exon. Additionally, it contained 17 novel ORFs (ORF1-4 and ORF6-18, Table 1) that were not present in other adenoviruses sequenced to date, nor did they match sequences in the NR protein database, using BLASTP and BLASTX. These unique ORFs encoded proteins of 33–312 amino acids (aa) in length (Table 1). Among these, the novel ORF8 was predicted to encode a 39 aa-length protein (molecular weight/theoretical isoelectric point, Mw/pI-4.49 kDa/8.96). This protein was predicted to contain a protein homolog to hepatitis E virus capsid protein (PDB: c3ggqA) by Phyre2 with a 74% sequence coverage, but the confidence of predicted protein structure by phyre2 was quite low as 40%. Therefore, there was no good structure predicted by using Phyre2 and SWISS-MODEL.
Four novel ORFs (ORF09, -10, -13, and -18) were predicted to contain transmembrane helices (TMHs), but no classical signal peptide. ORF10 was predicted to encode a 312 aa protein (Mw/pI-35.60 kDa/6.35) containing at least two TMHs. The orientation of the protein in the TMHs is shown in Figure 2. Furthermore, the TMHs detected by EMBOSS in Geneious also showed the presence of alpha-helices (α-helices) within TMHs predicted region, which was dominated by highly hydrophobic residues (red colour) (Figure 2A). However, we were unable to model the structure of ORF10 and or TMHs by using the Phyre2, HHpred, and SWISS-MODEL; this might be due to the lack of closely related structure in the database. Though the function of TMHs and dominant hydrophobic residues in this novel ORF is unknown, studies have shown that hydrophobicity drives the insertion of helical segments into the transmembrane proteins and acts as a hallmark of soluble globular protein tertiary structure [25][26]. ORF18 was predicted to encode a 237 aa protein (Mw/pI-26.14 kDa/8.81) that was also predicted to have at least two TMHs (Figure 3). TMHs detected in ORF18 by EMBOSS in Geneious also showed the presence of α-helices within TMHs predicted region, and they were also shown to be dominated by highly hydrophobic residues (red colour) (Figure 3A). Similarly, ORF13 was shown to contain a single TMH by TMHMM, TMpred, and EMBOSS tool in Geneious used in this study. The protein encoded by a novel ORF09 (181 aa) was predicted to have at least one C-terminal TMH by several programs, including EMBOSS 6.5.7 tool charge in Geneious, TMHMM and TMpred. However, there was an additional N-terminal TMH detected by Geneious and TMpred, and a further TMH was predicted by TMpred. Nonetheless, there was no evidence for conserved secondary structure and or protein homologs detected by various software, including HHpred [27], (Phyre2) [28], and SWISS-MODEL [29].
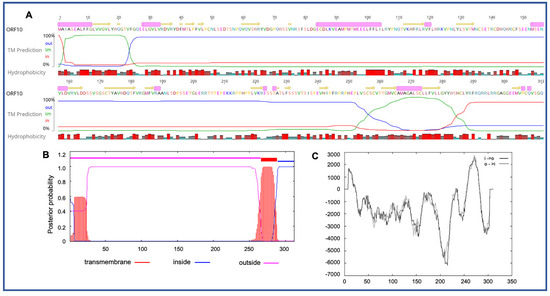
Figure 2. Predicted structure of the unique PaAdV1-ORF10. (A) prediction of transmembrane helices (TMHs) in unique PaAdV1-ORF10 gene using EMBOSS 6.5.7 tool in Geneious (version 10.2.2) (A), TMHMM (B), and TMpred (C). All the programs consistently predicted two TMHs. (A) TMHs detected by EMBOSS also showed the presence of alpha-helices within TMHs predicted region that has been dominated by highly hydrophobic residue (red colour). (B,C) The x-axis represents the position of residue, whereas y-axis represents the posterior probability (B), and scores (above 500 are considered significant) (C) for the predicted TMHs. (C) Solid and dashed black lines indicate protein orientation as inside to outside, and outside to inside, respectively.
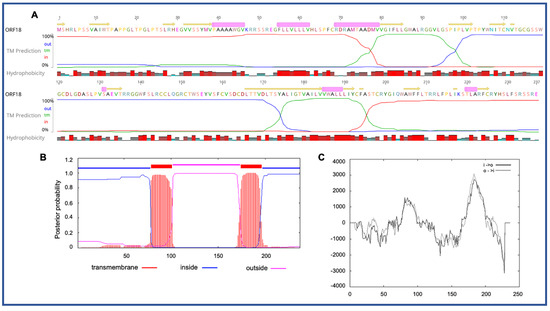
Figure 3. Predicted structure of the unique PaAdV1-ORF18. (A) prediction of transmembrane helices (TMHs) in unique PaAdV1-ORF18 gene, using EMBOSS 6.5.7 tool in Geneious (version 10.2.2) (A), TMHMM (B), and TMpred (C). All the programs consistently predicted two TMHs. (A) TMHs detected by EMBOSS also showed the presence of alpha-helices within TMHs predicted region that has been dominated by highly hydrophobic residue (red colour). (B,C) The x-axis represents the position of residue, whereas the y-axis represents the posterior probability (B) and scores (above 500 were considered significant) (C) for the predicted TMHs. (C) Solid and dashed black lines indicate protein orientation as inside to outside, and outside to inside, respectively.
5. Evolutionary Relationships of PaAdV-1
Phylogenetic reconstruction based on two non-structural (polymerase and pTP) and two structural (penton and hexon) protein sequences clearly supported the inclusion of the newly assembled PaAdV-1 in the genus Atadenovirus. In the resulting ML tree based on concatenated amino acid sequences of four selected AdVs genes, the novel PaAdV-1 occupied a distinct sub-clade position with strong bootstrap support, when compared with other sub-clades within the genus Atadenovirus (Figure 4), suggesting that it may represent an ancient evolutionary lineage within the genus. Using the same set of concatenated protein sequences, we found that the maximum inter-lineage sequence identity values between the novel PaAdV-1 and other atadenoviruses were >63% (PaAdV-1 vs. DAdV), >62.5% (PaAdV-1 vs. PsAdV-3), and >61.0% (PaAdV-1 vs. ruminant atadenoviruses), which mirrored the distinct phylogenetic position of this novel PaAdV-1. Furthermore, neighbour joining (NJ) phylogenetic inference of the concatenated protein sequences and the ML trees based on individual protein sequences of the complete polymerase, penton, and hexon genes demonstrated similar tree topologies for the representatives of atadenoviruses species. For example, all of these ML trees based on individual genes showed that the novel PaAdV-1 was placed phylogenetically into a distinct sub-clade from all other atadenoviruses (supported by a strong bootstrap) and did not show any obvious close evolutionary relationship. However, ML phylogeny based on the amino acid sequences of the complete pTP gene supported the closest relationship of novel PaAdV-1 with reptilian and bird atadenoviruses. Remarkably, in contrast to the previous studies where authors proposed the reptilian origin of atadenoviruses based on the comparison of phylogenetic trees of the adenoviruses, using a partial DNA polymerase gene [30][31], our resulting tress consistently demonstrated that atadenoviruses might have first evolved in a bird species that was present before the passerines and psittacine clades separated and was the ancestor of both clades (Figure 4).
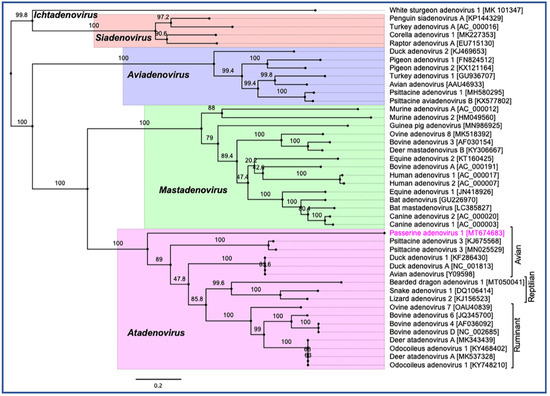
Figure 4. Phylogenetic tree shows the possible evolutionary relationship of novel passerine adenovirus 1 with other selected AdVs. Maximum likelihood (ML) tree was constructed by using concatenated amino acid sequences of the complete DNA-dependent DNA polymerase, pTP, penton, and hexon genes. Concatenated protein sequences were aligned with MAFTT (version 7.450) [32] in Geneious (version 10.2.2, Biomatters, Ltd., Auckland, New Zealand), under the BLOSUM62 scoring matrix and gap open penalty = 1.53. The gap >20 residues deleted from the alignments. The unrooted ML tree was constructed with PhyML [33] under the LG substitution model, and 1000 bootstrap re-samplings were chosen to generate ML trees, using tools available in Geneious (version 10.2.2, Biomatters, Ltd., Auckland, New Zealand). The numbers on the left show bootstrap values as percentages, and the labels at branch tips refer to original AdVs species name, followed by GenBank accession number in parentheses. The final tree is visualised with FigTree (version 1.4.4) [34]. The five official genera are highlighted as different background colours, and novel passerine adenovirus 1 is shown in pink colour.
References
- ICTV. Virus taxonomy, ninth report of the International Committee on Taxonomy of Viruses. In Virus Taxonomy; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier: San Diego, CA, USA, 2012; pp. 125–141.
- Benkö, M.; Harrach, B. A proposal for a new (third) genus within the family Adenoviridae. Arch. Virol. 1998, 143, 829–837.
- Dán, A.; Russell, W.C.; Ruzsics, Z.; Harrach, B.; Benkö, M. Analysis of the hexon gene sequence of bovine adenovirus type 4 provides further support for a new adenovirus genus (Atadenovirus). J. Gen. Virol. 1998, 79, 1453–1460.
- Vaz, F.F.; Raso, T.F.; E Agius, J.; Hunt, T.; Leishman, A.; Eden, J.-S.; Phalen, D.N. Opportunistic sampling of wild native and invasive birds reveals a rich diversity of adenoviruses in Australia. Virus Evol. 2020, 6.
- Pénzes, J.J.; Menéndez-Conejero, R.; Condezo, G.N.; Ball, I.; Papp, T.; Doszpoly, A.; Paradela, A.D.; Pérez-Berná, A.J.; López-Sanz, M.; Nguyen, T.H.; et al. Molecular Characterization of a Lizard Adenovirus Reveals the First Atadenovirus with Two Fiber Genes and the First Adenovirus with Either One Short or Three Long Fibers per Penton. J. Virol. 2014, 88, 11304–11314.
- Farkas, S.L.; Benkő, M.; Élő, P.; Ursu, K.; Dán, Á.; Ahne, W.; Harrach, B. Genomic and phylogenetic analyses of an adenovirus isolated from a corn snake (Elaphe guttata) imply a common origin with members of the proposed new genus Atadenovirus The GenBank accession number of the sequence reported in this paper is AY082603. J. Gen. Virol. 2002, 83, 2403–2410.
- Harrach, B. Adenoviruses: General Features. In Reference Module in Biomedical Sciences; Elsevier: Amsterdam, The Netherlands, 2014.
- Vrati, S.; Brookes, D.; Strike, P.; Khatri, A.; Boyle, D.; Both, G. Unique Genome Arrangement of an Ovine Adenovirus: Identification of New Proteins and Proteinase Cleavage Sites. Virology 1996, 220, 186–199.
- Miller, M.M.; Cornish, T.E.; Creekmore, T.E.; Fox, K.; Laegreid, W.; McKenna, J.; Vasquez, M.; Woods, L.W. Whole-genome sequences of Odocoileus hemionus deer adenovirus isolates from deer, moose and elk are highly conserved and support a new species in the genus Atadenovirus. J. Gen. Virol. 2017, 98, 2320–2328.
- Pénzes, J.J.; Szirovicza, L.; Harrach, B. The complete genome sequence of bearded dragon adenovirus 1 harbors three genes encoding proteins of the C-type lectin-like domain superfamily. Infect. Genet. Evol. 2020, 83, 104321.
- To, K.K.-W.; Tse, H.; Chan, W.-M.; Choi, G.K.Y.; Zhang, A.J.X.; Sridhar, S.; Wong, S.C.Y.; Chan, J.F.; Chan, A.S.F.; Woo, P.C.Y.; et al. A Novel Psittacine Adenovirus Identified During an Outbreak of Avian Chlamydiosis and Human Psittacosis: Zoonosis Associated with Virus-Bacterium Coinfection in Birds. PLOS Neglected Trop. Dis. 2014, 8, e3318.
- Duarte, M.A.; Silva, J.M.F.; Brito, C.R.; Teixeira, D.S.; Melo, F.; Ribeiro, B.M.; Nagata, T.; Campos, F.S. Faecal Virome Analysis of Wild Animals from Brazil. Viruses 2019, 11, 803.
- Needle, D.B.; Wise, A.G.; Gregory, C.R.; Maes, R.K.; Sidor, I.F.; Ritchie, B.W.; Agnew, D. Necrotizing Ventriculitis in Fledgling Chimney Swifts (Chaetura Pelagica) Associated With a Novel Adenovirus, Chimney Swift Adenovirus-1 (CsAdV-1). Veter. Pathol. 2019, 56, 907–914.
- De Oliveira, A.P.J.; Rangel, M.C.V.; Vidovszky, M.Z.; Rossi, J.L., Jr.; Vicentini, F.; Harrach, B.; Kaján, G.L. Identification of two novel adenoviruses in smooth-billed ani and tropical screech owl. PLoS ONE 2020, 15, e0229415.
- Harrach, B.; Meehan, B.M.; Benkő, M.; Adair, B.M.; Todd, D. Close Phylogenetic Relationship between Egg Drop Syndrome Virus, Bovine Adenovirus Serotype 7, and Ovine Adenovirus Strain 287. Virology 1997, 229, 302–306.
- Hess, M.; Blöcker, H.; Brandt, P. The Complete Nucleotide Sequence of the Egg Drop Syndrome Virus: An Intermediate between Mastadenoviruses and Aviadenoviruses. Virology 1997, 238, 145–156.
- Phalen, D.N.; Agius, J.; Vaz, F.F.; Eden, J.-S.; Setyo, L.C.; Donahoe, S. A survey of a mixed species aviary provides new insights into the pathogenicity, diversity, evolution, host range, and distribution of psittacine and passerine adenoviruses. Avian Pathol. 2019, 48, 437–443.
- Rinder, M.; Schmitz, A.; Baas, N.; Korbel, R. Molecular identification of novel and genetically diverse adenoviruses in Passeriform birds. Virus Genes 2020, 56, 316–324.
- Karen, A.F.; Levi, A.; Laura, H.-H.; Myrna, M. A Mortality Event in Elk (Cervus elaphus) Calves Associated with Malnutrition, Pasteurellosis, and Deer Adenovirus in Colorado, USA. J. Wildl. Dis. 2017, 53, 674–676.
- Thomson, D.; Meers, J.; Harrach, B. Molecular confirmation of an adenovirus in brushtail possums (Trichosurus vulpecula). Virus Res. 2002, 83, 189–195.
- Gál, J.; Mándoki, M.; Sós, E.; Kertész, P.; Koroknai, V.; Banyai, K.; Farkas, S.L. Novel adenovirus detected in kowari (Dasyuroides byrnei) with pneumonia. Acta Microbiol. et Immunol. Hung. 2017, 64, 81–90.
- Garcia-Morante, B.; Pénzes, J.J.; Costa, T.; Martorell, J.; Martinez, J. Hyperplastic stomatitis and esophagitis in a tortoise (Testudo graeca) associated with an adenovirus infection. J. Veter. Diagn. Investig. 2016, 28, 579–583.
- Fu, G.; Chen, H.; Yu, H.; Cheng, L.; Fu, Q.; Shi, S.; Wan, C.; Chen, C.; Lin, J. Full Genome Sequence of Egg Drop Syndrome Virus Strain FJ12025 Isolated from Muscovy Duckling. Genome Announc. 2013, 1, e00623-13.
- Davison, A.J.; Benkő, M.; Harrach, B. Genetic content and evolution of adenoviruses. J. Gen. Virol. 2003, 84, 2895–2908.
- Silverman, B.D. Hydrophobicity of transmembrane proteins: Spatially profiling the distribution. Protein Sci. 2003, 12, 586–599, doi:10.1110/ps.0214903.
- Elazar, A.; Weinstein, J.J.; Prilusky, J.; Fleishman, S.J. Interplay between hydrophobicity and the positive-inside rule in determining membrane-protein topology. Proc. Natl. Acad. Sci. USA 2016, 113, 10340–10345, doi:10.1073/pnas.1605888113.
- Zimmermann, L.; Stephens, A.; Nam, S.-Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J. Mol. Boil. 2018, 430, 2237–2243, doi:10.1016/j.jmb.2017.12.007.
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858, doi:10.1038/nprot.2015.053.
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; Beer, T.A.P.D.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303, doi:10.1093/nar/gky427.
- Wellehan, J.F.X.; Johnson, A.J.; Harrach, B.; BenköM.; Pessier, A.P.; Johnson, C.M.; Garner, M.M.; Childress, A.; Jacobson, E.R. Detection and Analysis of Six Lizard Adenoviruses by Consensus Primer PCR Provides Further Evidence of a Reptilian Origin for the Atadenoviruses. J. Virol. 2004, 78, 13366–13369, doi:10.1128/jvi.78.23.13366-13369.2004.
- Harrach, B.; Information, R. Reptile Adenoviruses in Cattle? Acta Veter. Hung. 2000, 48, 485–490, doi:10.1556/004.48.2000.4.11.
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780.
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Boil. 2010, 59, 307–321, doi:10.1093/sysbio/syq010.
- Molecular evolution, phylogenetics and epidemiology. FigTree. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 2 December 2011).

