Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Junjie Luo | -- | 3823 | 2022-06-24 02:31:12 | | | |
| 2 | Conner Chen | + 4 word(s) | 3827 | 2022-06-24 05:00:20 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Luo, J. Mitochondrial Ca2+ and Tumor Cell. Encyclopedia. Available online: https://encyclopedia.pub/entry/24420 (accessed on 12 June 2026).
Luo J. Mitochondrial Ca2+ and Tumor Cell. Encyclopedia. Available at: https://encyclopedia.pub/entry/24420. Accessed June 12, 2026.
Luo, Junjie. "Mitochondrial Ca2+ and Tumor Cell" Encyclopedia, https://encyclopedia.pub/entry/24420 (accessed June 12, 2026).
Luo, J. (2022, June 24). Mitochondrial Ca2+ and Tumor Cell. In Encyclopedia. https://encyclopedia.pub/entry/24420
Luo, Junjie. "Mitochondrial Ca2+ and Tumor Cell." Encyclopedia. Web. 24 June, 2022.
Copy Citation
Mitochondrial Ca2+ transport-related processes are involved in important biological processes of tumor cells including proliferation, metabolism, and apoptosis. In particular, MCU and its regulatory proteins represent a new era in the study of MCU-mediated mitochondrial Ca2+ homeostasis in tumors.
mitochondrial calcium
calcium homeostasis
calcium regulation
tumor
1. Mitochondrial Ca2+ and Energy Metabolism of Tumor Cells
Ca2+ participates in almost all physiological activities in cells. Mitochondria were originally considered to be a “Ca2+ pool” with the ability to absorb a large amount of Ca2+, and the uptake of Ca2+ by mitochondria increases significantly when the extramitochondrial Ca2+ is overloaded [1]. It has been found that Ca2+ can stimulate glycogen decomposition and glucose oxidation, resulting in an increase in ATP supply [2]. The increase in cytoplasmic Ca2+ concentration is transmitted to mitochondria and Ca2+-activated dehydrogenase is a key rate control enzyme in the tricarboxylic acid cycle (TAC) flux. Ca2+ activation will lead to the increase in pyridine nucleotide reduction and oxidative phosphorylation [3]. Mitochondrial Ca2+ uptake can activate matrix enzymes, stimulate ATP production, and regulate energy metabolism by activating pyruvate dehydrogenase, isocitrate dehydrogenase, and ketoglutarate dehydrogenase. This “parallel activation model” provides a mechanism in which Ca2+ stimulates the process of energy consumption caused by physiological activities such as various hormones, muscle contraction, or increased cardiac load [4]. It also provides a means for cells to upregulate ATP supply to keep up with this energy consumption.
Metabolic reprogramming in tumor cells is considered to be a sign of cancer and is involved in tumor growth and development. Compared to normal cells, under the condition of sufficient aerobic supply, tumor cells still obtain energy by aerobic glycolysis and produce a large amount of lactic acid and a small amount of ATP [5]. M2 isoform of pyruvate kinase (PKM2) is critical for the metabolic fate of the glycolytic intermediates [6][7][8]. During the course of the disease, tumor cells will develop overall metabolic adaptability so that they can survive in the tumor microenvironment with low oxygen and nutrient levels [9].
In summary, Ca2+ affects the functional changes of mitochondria (such as mitochondrial dysfunction, metabolic conversion to glycolysis, and mtDNA mutations) and thus, cell energy metabolism, which is closely related to the occurrence and development of tumors (Figure 1). At present, the adaptability of tumor cell metabolism is the main limitation of cancer treatment, which is highly related to the resistance to therapeutic drugs [10]. The unique metabolic pattern of tumor cells is both a challenge and an opportunity. Understanding the metabolic mechanism of tumor cells is greatly significant for the early diagnosis of a tumor’s metabolic phenotype and rational targeted therapy.
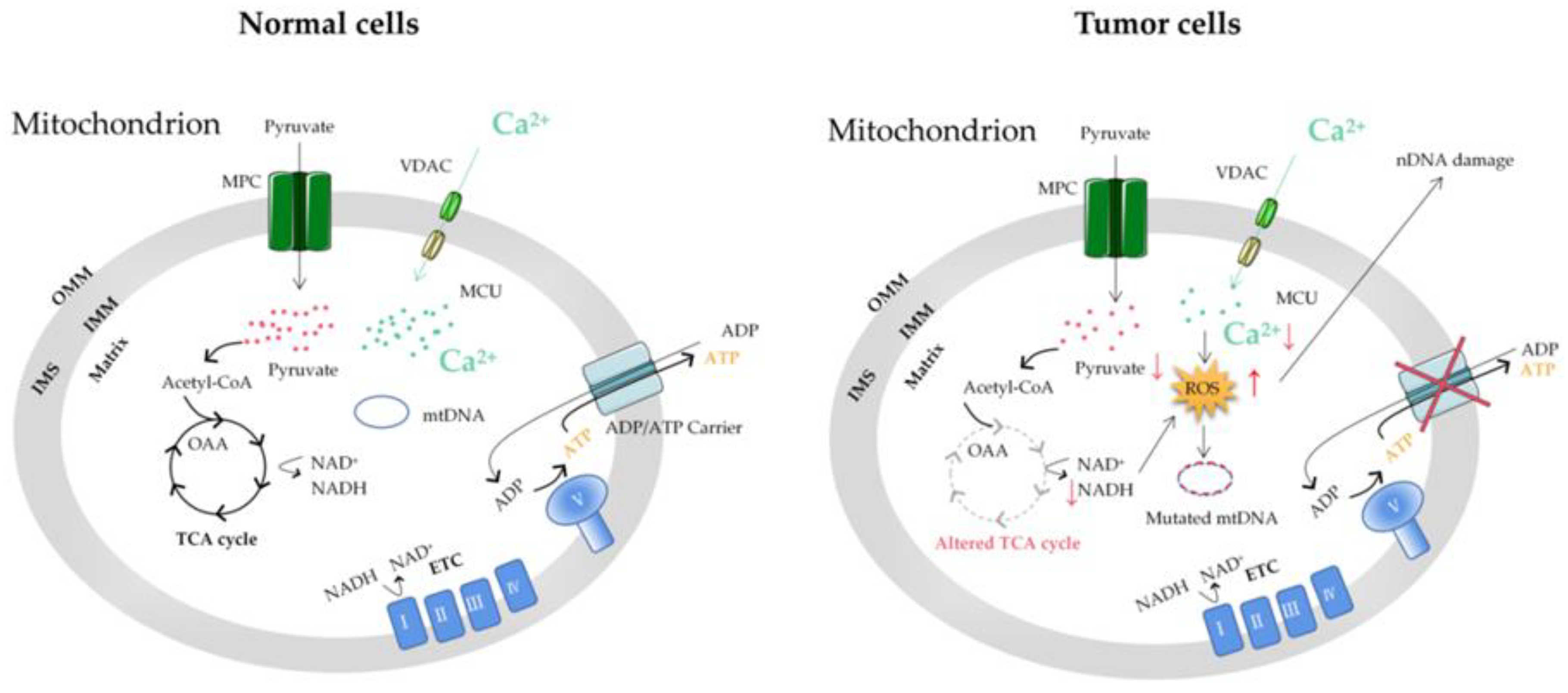
Figure 1. The mitochondrial Ca2+ and energy metabolism in normal and tumor cells. The reprogramming of energy metabolism, including energy production disorders caused by cell respiratory defects, is the core symbol of cancer. The change in energy metabolism in cancer cells is related to the abnormal function of mitochondria. Accumulation of the ROS induced by mitochondrial Ca2+ dyshomeostasis and altered TCA in tumor cells can cause mitochondrial DNA (mtDNA) and nuclear DNA (nDNA) mutations. MCU, mitochondrial calcium uniporter; VDAC, voltage-dependent anion-selective channel; ETC, electron transport chain; OMM, outer mitochondrial membrane; IMS, intermembrane space; IMM, inner mitochondrial membrane; MPC, mitochondrial pyruvate carrier; OAA, oxaloacetic acid; TCA cycle, tricarboxylic acid cycle.
2. Mitochondrial Ca2+ and the MCU in Autophagy/Mitophagy of Tumor Cells
Metabolic adaptations allow tumor cells to survive in the low oxygen and nutrient tumor microenvironment. Among these metabolic adaptations, tumor cells use glycolysis but also mitochondrial oxidation to generate ATP; another particular adaptation of tumor cell metabolism is the use of autophagy and mitophagy [11]. Autophagy plays a key role in maintaining cellular homeostasis [12]. Thus, autophagy disorders disrupt normal physiological processes and are implicated in the pathogenesis of various diseases, including tumors [13]. Autophagy is a highly conserved catabolic process that results in the degradation and recycling of proteins and organelles after the fusion of isolated vesicles, autophagosomes, and lysosomes that provide hydrolases [14]. The molecular process of autophagy is complex and involves sequential steps for nucleation, extension, and fusion of associated proteins, including autophagy-associated proteins [15]. Autophagy has two main physiological roles: the breakdown of dysfunctional proteins or organelles as a quality control mechanism and the recovery of biological macromolecules to maintain metabolic needs under nutritional stress [16]. Autophagy has been found to play two roles in a tumor: a protective role in the early stages of the tumor and the promotion of tumor growth in advanced stages [17].
Intracellular Ca2+ is considered a bidirectional regulator of autophagy [18][19], which may depend on the spatiotemporal parameters of Ca2+ signal transduction, nutrients, and the utilization of growth factors [20]. Ca2+ overload can affect autophagy, leading to normal cell carcinogenesis and the growth of tumor cells. It is demonstrated that Ca2+ agonists, such as vitamin D3 compounds, ionomycin, ATP, and thapsigargin, can stimulate the autophagy of MCF-7 breast tumor cells through Ca2+-activated kinase CaMKK [21]. Consistent with the activation of autophagy by Ca2+, researchers have found that mitochondrial fission-mediated Ca2+ signaling also significantly induces autophagy in HCC [22]. Conversely, some other research groups have found the inhibitory effect of Ca2+ on autophagy. At present, there are several ways for Ca2+ to inhibit autophagy: (1) the inositol 1,4,5-trisphosphate receptor (IP3R) mediates Ca2+ to reduce the release of Beclin1 so as to reduce autophagosome production and inhibit autophagy; (2) IP3R mediates Ca2+ activation of calpain, separates autophagy protein 5 from autophagy protein 12, reduces the level of their complex, and inhibits autophagy [23]; (3) The increase of Ca2+ released by the ER to the mitochondria enhances the TAC and ATP production and inhibits autophagy [24][25]; (4) IP3R mediates Ca2+ into mitochondria, resulting in increased ATP production and the inhibition of AMPK, thereby inhibiting autophagy. Therefore, Ca2+ may have different regulatory effects on autophagy.
As with non-selective autophagy, the role of mitophagy is complex and can depend on tumor type and stage. Since both autophagy and mitophagy are related to mitochondrial function, targeting mitochondrial ion channels may also be an interesting strategy to regulate autophagy or mitophagy in tumors. Ca2+ exchanges have been associated with autophagy and mitophagy regulation. Therefore, unsurprisingly, some mitochondrial calcium transporters, such as the MCU, have recently been found to be involved in autophagy and mitophagy regulation in tumor cells.
The MCU is generally considered to be the main Ca2+ transporter in the matrix, which is a major mediator of calcium influx into mitochondria. The MAM is an important part of Ca2+ transfer from the ER to the mitochondria to regulate mitochondrial enzymes. Ca2+ flow mainly occurs through IP3R and transient receptor potential cation channel subfamily M member 8 (TRPM8) in the ER membrane [26]. Sensitizing IP3Rs and the interruption of Ca2+ flow between the ER and the mitochondria break the calcium homeostasis and decrease mitochondrial bioenergetics, which subsequently decreases OXPHOS and activates autophagy [27][28]. However, unlike normal cells, autophagy activation caused by MAM destruction in tumor cells seems insufficient to maintain the required energy level, resulting in tumor cell death and reduced tumor growth [29] (Figure 2). Although the mechanisms linked with autophagy are not clearly understood, the MCU could be an interesting target to disrupt Ca2+ in the MAM in tumor cells, decreasing mitochondrial function and inducing cell death. The MCU has also been found to be altered in tumors from different tissues [30]. In particular, the expression of the MCU is associated with tumor progression and metastasis [31]. Therefore, mitochondrial Ca2+ and the MCU represent attractive antitumor targets for regulating mitochondrial dysfunction and autophagy/mitophagy in tumors.
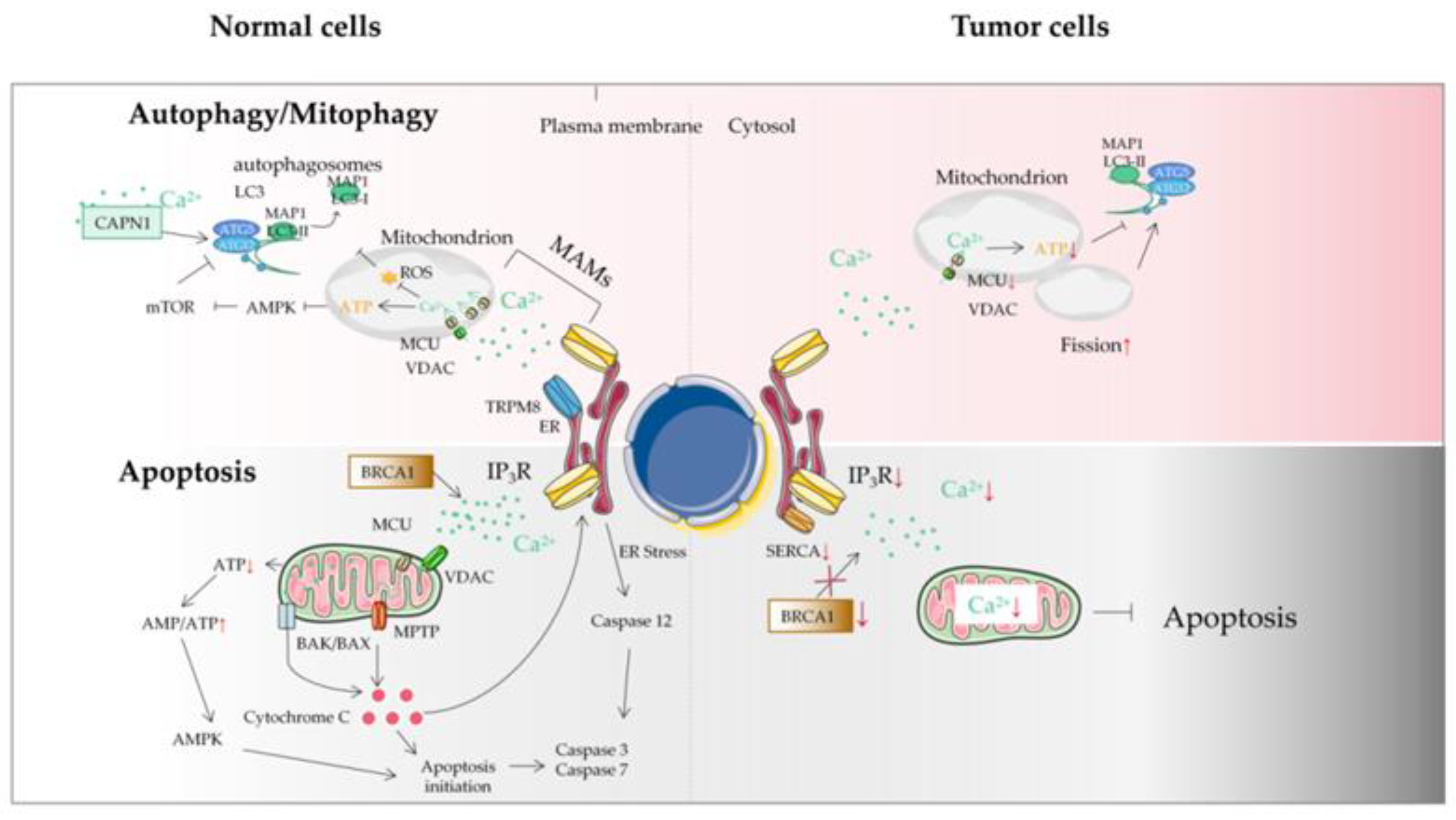
Figure 2. The autophagy/mitophagy and apoptosis of tumor cells. Autophagy plays a key role in maintaining cellular homeostasis. Autophagy disorder destroys normal physiological processes and can lead to cancer. Ca2+ can inhibit autophagy through an IP3R- or ER-mediated manner. Some mitochondrial Ca2+ transporters are also involved in autophagy and mitophagy regulation. Autophagy plays two roles in a tumor: a protective role in the early stages of tumor and the promotion of tumor growth in advanced stages. Tumor cells may avoid apoptosis by reducing Ca2+ influx into the cytoplasm. It can be achieved by downregulation of the expression of Ca2+ channels in the plasma membrane or by reducing the effectiveness of the signal pathways that activate these channels. This protective measure will greatly reduce the response of Ca2+ overload to pro-apoptotic stimulus, thus impairing the effectiveness of mitochondrial and cytoplasmic apoptotic pathways in tumor cells. Another mechanism is that tumor cells adapt to the reduction of Ca2+ in ER, without inducing the pro-apoptotic ER stress response usually accompanied by ER Ca2+ imbalance. ER, endoplasmic reticulum; MAM, mitochondrial-associated ER membrane; MCU, mitochondrial calcium uniporter; MPTP, mitochondrial permeability transition pore; ROS, reactive oxygen species; TRPM8, transient receptor potential cation channel subfamily M member 8; VDAC, voltage-dependent anion-selective channel; SERCA, sarco-endoplasmic reticulum Ca2+-ATPase; IP3R, inositol triphosphate receptor; BRCA1, breast cancer susceptibility gene.
3. Mitochondrial Ca2+ and Tumor Cell Apoptosis
Apoptosis involves the activation, expression, and regulation of a series of genes. It is not a phenomenon of autologous injury under pathological conditions, but a death process actively striving for better adaptation to the living environment [32]. The regulation of apoptosis is controlled by a very complex signal network system. There are three major signaling pathways: the mitochondrial pathway, the death receptor pathway, and the ER pathway [33]. These signal transduction pathways can eventually activate caspase-3, the executor of apoptosis, which hydrolyzes various cellular components and causes cell apoptosis [34]. In animal cells, the mitochondrial pathway is the most common apoptotic mechanism and the core of apoptosis [35][36]. In the early stage of apoptosis, mitochondria show changes such as increased permeability, Ca2+ uptake, decreased transmembrane potential, and the release of cytochrome C and apoptosis-inducing factors [37]. Changes in Ca2+ concentration may play a key role in the early apoptotic signal transduction pathway upstream of mitochondria [38]. However, this sensitive system can be affected to drive malignant transformation in cells.
In the process of apoptosis, intracellular Ca2+ overload can come from either extracellular Ca2+ influx or the release of the intracellular Ca2+ pool [39]. Some studies have suggested that the release of the intracellular Ca2+ pool can only cause a temporary increase in Ca2+, which is not enough to cause apoptosis. The triggering of apoptosis requires Ca2+ to reach a certain threshold and maintain this level for a long time [40]. With further research, there are many factors regulating Ca2+ levels in mitochondria, including intracellular regulation of the Bcl-2 family, the release of calcium pool ER and the participation of ROS [41]. At present, more than 20 members of the Bcl-2 family have been found. The proteins in the Bcl-2 family are widely distributed in the outer membrane of the mitochondria, nuclear membrane, and ER, regulating the activity of the caspases. Bcl-2 family members can be divided into three groups according to their structure and function. The first group includes Bcl-2, Bcl-XL, and Bcl-W, which have anti-apoptotic properties. The second group is a member of the Bcl-2 family with BH3-only proteins, which could increase the permeability of the mitochondrial outer membrane during cell apoptosis [42]. The third group, which contains all the domains except BH4, also increases membrane permeability and has pro-apoptotic activity [43].
The ER is an important Ca2+ reservoir in eukaryotic cells, so Ca2+ in the ER must maintain a stable level to ensure the accuracy of the Ca2+ signal [44]. Ca2+ released from the ER can directly flow into mitochondria and the uptake rate of mitochondrial Ca2+ depends on the concentration gradient of cytoplasmic Ca2+ at the IP3R opening on the ER. The opening of the ER InsP3/Ca2+ channel affects the Ca2+ balance in mitochondria and the InsP3/Ca2+ channel is one of the targets of caspase-3 [45]. Moreover, ER stress induced by the disturbance in the ER calcium state can activate caspase-12, a specific ER-localized protein, to trigger apoptosis in a mitochondria-independent way [46][47].
Mitochondria are the central link mediating apoptosis, as well as the main site of ROS generation [48]. With the discovery and further understanding of the role of mitochondrial Ca2+ in apoptosis, the research on the role of ROS in apoptosis is getting more and more in-depth. The regulation mechanisms of ROS on mitochondrial Ca2+ homeostasis are as follows: (1) After cells receive the pro-apoptotic signals, the increase of ROS promotes mitochondrial Ca2+ influx, which may be caused by affecting voltage-dependent Ca2+ channels, non-specific cell membrane Ca2+ permeability changes, and Na+/Ca2+ exchanges [49]; (2) Increased intracellular Ca2+ can activate other enzymes to further upregulate the level of oxygen free radicals, so ROS can indirectly produce more oxides and further promote the rise in mitochondrial Ca2+ level [50]. In addition, an overload of Ca2+ leading to oxidative metabolism impairment and ROS overproduction [51]. Previous studies have suggested that under oxygen stress, ROS produced by mitochondria will cause membrane lipid peroxidation and changes in mitochondrial function, resulting in the release of Ca2+ and apoptosis of mitochondria [52]; (3) ROS can regulate IP3R production and affect Ca2+ release from the ER into mitochondria [53]; (4) ROS can also affect the sarcoplasmic reticulum Ca2+ pump and inhibit intracellular or extracellular ER Ca2+ transfer by inhibiting the Ca2+-ATPase pump [54]; (5) Both ROS and Ca2+ can induce MPTP opening. On the other hand, MPTP opening leads to a large increase in ROS [55].
Several types of tumor cells have experienced extensive reorganization of their internal Ca2+ signal transduction mechanism, which promotes the occurrence of tumors [56]. Calcium ion exchange between mitochondria and the ER can be carried out through some Ca2+ signal proteins, including VDAC1, IP3R, and SERCA, which play vital roles in the processes of tumors. VDAC1 plays a significant role in cellular Ca2+ homeostasis and it has also been recognized as a key protein in mitochondria-mediated apoptosis [57]. For example, in several types of non-small cell lung cancer and cervical cancer, the expression level of VDAC1 is related to tumor growth and invasion [58]. The downregulation of IP3R1 in bladder cancer cells prevents mitochondrial Ca2+ overload by decreasing the uptake of ER−mitochondria Ca2+, thereby reducing cisplatin-mediated apoptosis [59]. The significant reduction or loss of SERCA3 subtypes in transformed colonic epithelial cells also proves that the Ca2+ signal is remodeled in tumorigenesis [60].
Recently, it has been found that in several cancer types, the imbalance of two new mechanisms will affect the renewal of the proteasome, so as to regulate the apoptosis sensitivity of tumor cells by affecting IP3R3 proteins and interfering with the Ca2+ exchange between the ER and mitochondria [61]. (1) The tumor suppressor protein PTEN and F-box/LRR repeat protein 2 (FBXL-2) compete for binding to IP3R3, which slows down FBXL-2-mediated IP3R3 proteasome degradation. This represents a new mechanism. The deletion of PTEN enables tumor cells to avoid apoptosis [62]. The downregulation of IP3R3 impairs the pro-apoptotic mitochondrial Ca2+ transfer. (2) The tumor suppressor protein BRCA1-associated protein 1 (BaP1) is a deubiquitinase that promotes the transfer of ER−mitochondria Ca2+ by stabilizing IP3R3. Under long-term environmental pressure, the function of BaP1 will be seriously disrupted, which is related to the acquired inactivating mutations of the BaP1 gene. The loss of BaP1 will lead to the downregulation of IP3R3, which hinders the effective apoptotic clearance of damaged cells and is conducive to the occurrence of tumors and the survival of malignant cells [63]. In addition, oncogenes and tumor suppressor proteins can play other roles in cancer development through Ca2+ signal regulation, such as resistance to apoptosis. Because mitochondrial Ca2+ overload is related to apoptosis and death, modifying ER−mitochondria Ca2+ transfer at the MAM will change the sensitivity of apoptosis, and tumor cells can acquire resistance to cell death accordingly [64]. For example, by inhibiting IP3R-mediated Ca2+ signaling or increasing the transmembrane distance at the MAM, the efficiency of ER−mitochondria Ca2+ transfer can be reduced, so as to decrease the sensitivity of tumor cells to apoptosis [65] (Figure 2).
4. The Relationship between the MCU and the Tumor
With the deepening of the research on the mechanism of cancer metastasis, the relationship between mitochondrial calcium homeostasis and the development of malignant tumors has attracted much attention [66][67]. The MCU is a major mediator of calcium influx into mitochondria and controls cellular energy metabolism, autophagy/mitophagy, and apoptosis. In most cancer tissues, the MCU showed moderate to strong immunostaining [68]. Increasingly, evidence shows that the MCU is closely related to multiple cancers, such as breast cancer, HCC, and colon cancer.
The MCU plays an important role in controlling the energy metabolism of tumor cells. The receptor-interacting protein kinase 1 (RIPK1) is an important signal molecule in the pathway of cell survival, apoptosis, and necrosis, which is significantly upregulated in colorectal cancer (CRC) cells. RIPK1 interacts with the MCU to promote CRC cell proliferation by increasing mitochondrial Ca2+ uptake and energy metabolism [69]. Compared with normal tissues, the MCU, MICU1, and MICU2 were overexpressed in oral squamous cell carcinoma (OSCC) tissues. The MCU is a new proto-oncogene of OSCC, which is regulated by nuclear factor erythroid 2-related factor 2 (Nrf2) transcription. The MCU can enhance the proliferation of OSCC cells and inhibit apoptosis [70]. Dihydroartemisinin can repress the proliferation and migration of OSCC cells by inhibiting the expression of the MCU [71]. MCU-mediated high mitochondrial Ca2+ can increase the proliferation of prostate cancer cells by inhibiting MPTP [72]. The MCU is involved in the autophagy of cancer cells. In kidney cancer cells, the upregulation of miR501-5p leads to the downregulation of the MCU, which leads to the activation of AMPK, thus promoting mTOR-independent autophagy [73]. The MCU also affects the apoptosis of cancer cells. Cathepsin S (CTSS) is overexpressed in glioblastomas (GBs). High levels of CTSS are associated with tumor progression and a poor prognosis of GB. Inhibiting the expression of CTSS in GB cells can increase the expression of MCUs. Enhanced mitochondrial Ca2+ uptake leads to mitochondrial Ca2+ overload, produces a large number of ROS, and, finally, causes apoptosis [74]. RY10-4 can induce the apoptosis of breast cancer cells by elevating Ca2+ through the MCU [75]. The MCUR1 is frequently upregulated in HCC cells, which enhances Ca2+ uptake into mitochondria in an MCU-dependent manner. The HCC cell survival rate is significantly improved by inhibiting mitochondrial-dependent apoptosis and promoting HCC cell proliferation, resulting in poor prognosis [76]. The data also show that miR-25 decreases mitochondrial Ca2+ uptake through selective MCU downregulation, thereby reducing apoptosis. The MCU seems to be downregulated in human colon cancer samples. Correspondingly, miR-25 is abnormally expressed, indicating that mitochondrial Ca2+ plays an important role in the survival of cancer cells [77].
Current studies have suggested that the MCU is correlated with tumor size and lymphatic infiltration, which may contribute to tumor growth and metastasis [78][79]. It is speculated that the MCU affects the expression of VEGF through HIF-1α and the inhibition of MCU expression significantly reduces the invasion and migration ability of breast cancer cells [80][81]. In addition, the expression of the MCUR1 significantly affects the progression and prognosis of breast cancer [82][83]. However, the role of the MCU in cancer research remains controversial. Studies have shown that specific Ca2+ channels play different roles in some cancers due to different regulatory mechanisms. Previous studies have revealed that a highly expressed MCU promotes the metastasis of adrenocortical carcinoma breast cancer cells with poor prognosis. In hepatocellular carcinoma studies, MCU-dependent mitochondrial Ca2+ uptake promotes metastasis of HCC cells [84]. In fact, it can be analyzed the transcriptional expression levels of MCUs in different cancers through the relevant database (https://tnmplot.com/analysis/; accessed on 31 March 2022) [85] and demonstrated that the expression levels of MCUs in most tumors are not consistent with those in normal tissues (Figure 3). The majority of tumors have significantly elevated levels of MCU expression. However, high MCU expression in cancer patients may not always be beneficial. Coincidentally, it can be analyzed the survival curves between MCU expression levels and cancer patient survival through the GEPIA database (http://gepia.cancer-pku.cn/index.html; accessed on 31 March 2022) and found that in adrenocortical carcinoma and hepatocellular carcinoma, overall survival is significantly greater in low MCU expression than in high MCU expression (Figure 4A,B). On the contrary, in renal clear cell carcinoma and brain lower grade glioma, overall survival is significantly greater in high MCU expression than in low MCU expression (Figure 4C,D). To sum up, the relationship between the MCU and tumor is complex and needs more in-depth research.
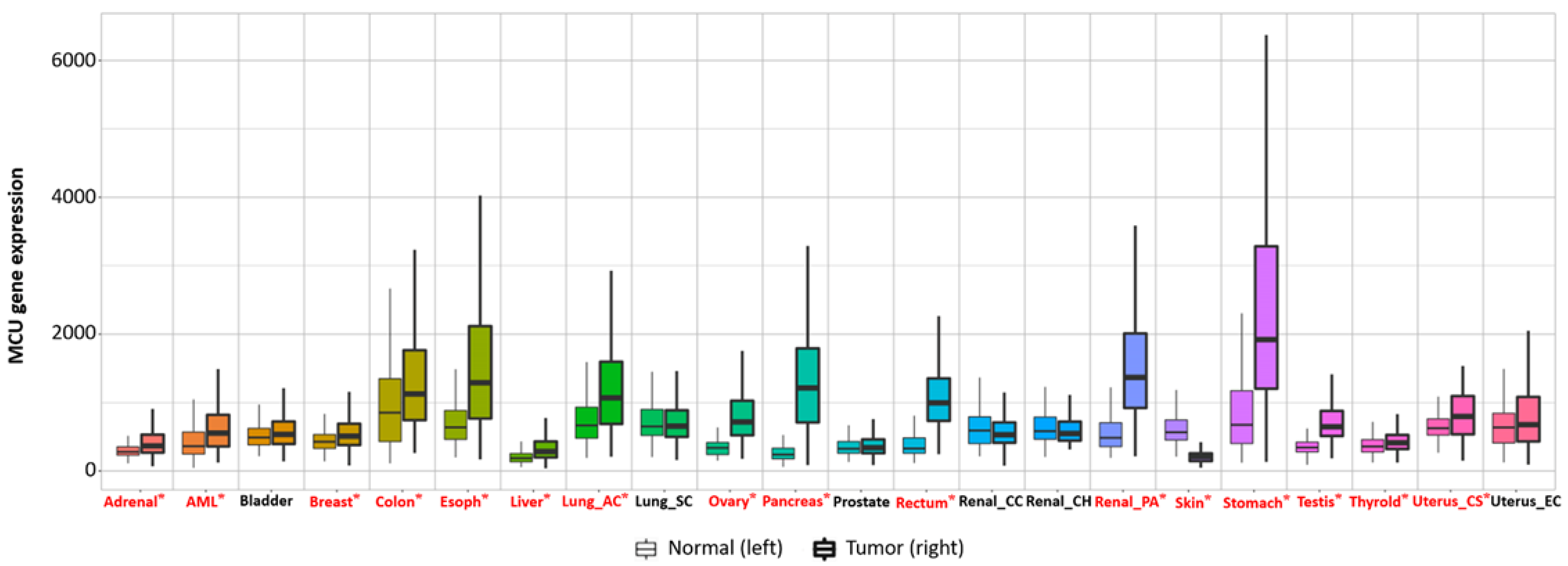
Figure 3. Transcriptional expression level of the MCU in various normal and cancerous organs. The MCU is closely related to multiple cancers; the expression levels of the MCU in most tumors are not consistent with those in normal tissues. The majority of tumors have significantly elevated levels of MCU expression. Significant differences by Mann−Whitney U test are marked with red and *. MCU, mitochondrial calcium uniporter.
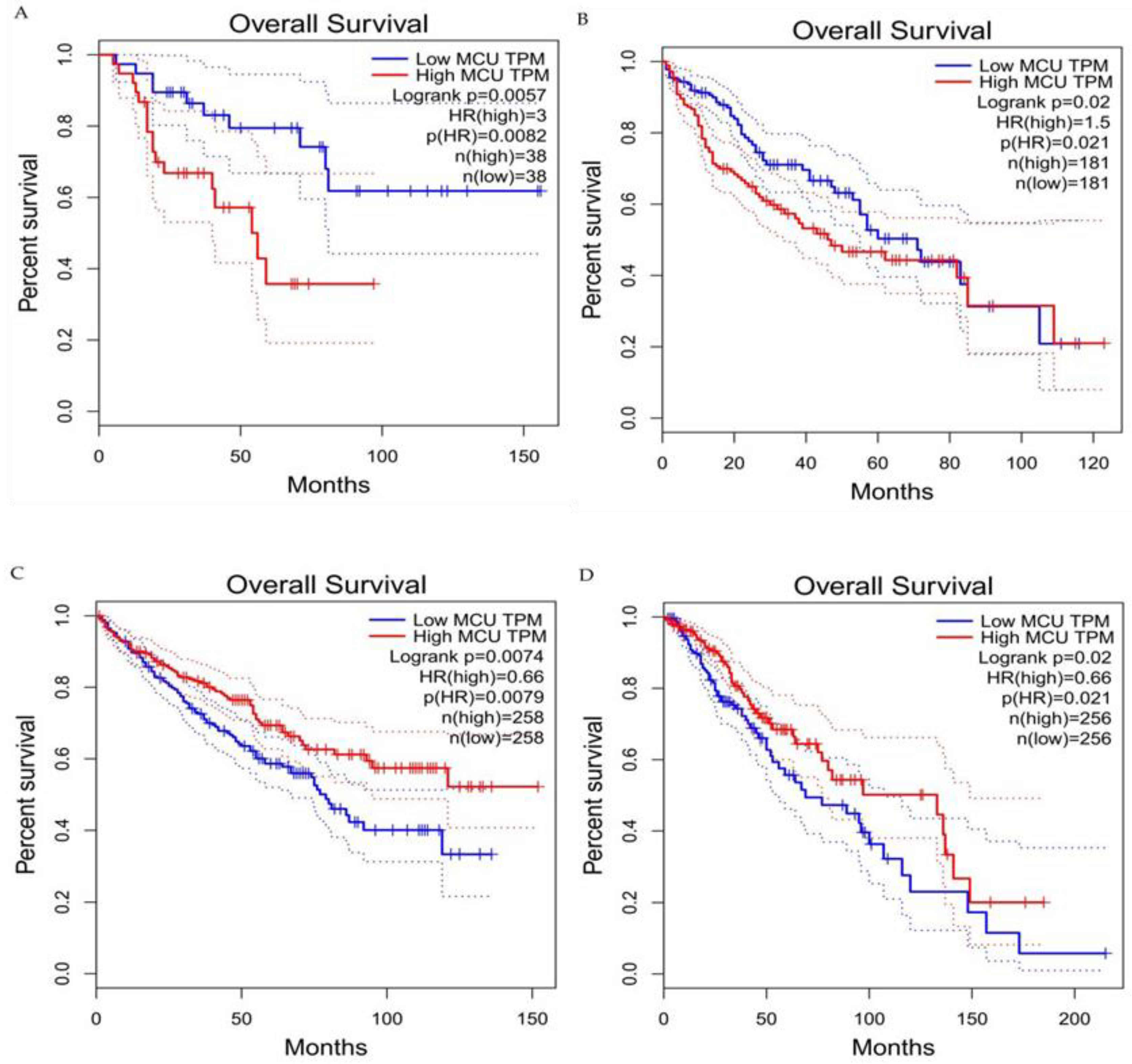
Figure 4. Survival curves for overall survival of high versus low expressing MCU. (A) Adrenocortical carcinoma. (B) Hepatocellular carcinoma. (C) Renal clear cell carcinoma. (D) Brain lower grade glioma. In adrenocortical carcinoma and hepatocellular carcinoma, overall survival is significantly greater in low MCU expression than in high MCU expression. In renal clear cell carcinoma and brain lower grade glioma, overall survival is significantly greater in high MCU expression than in low MCU expression. MCU, mitochondrial calcium uniporter; HR, hazard rate.
Mitochondria regulate Ca2+ homeostasis through the uptake of Ca2+ into the mitochondria via MCU and the release of Ca2+ from the mitochondria via NCLX, regulating intramitochondrial and intracytoplasmic Ca2+ concentrations. Therefore, the regulation of both is deeply intertwined. Since the reorganization of cytosolic calcium signaling commonly occurs in tumor cells, mitochondrial calcium imbalance causes alterations in cytosolic calcium signaling and thus, affects tumorigenesis and progression [86]. Given the important impact of mitochondrial calcium imbalance on tumors, a large number of studies have used mitochondrial calcium imbalance as a starting point to explore new diagnostic and therapeutic approaches to tumors. It is found that proteins associated with mitochondrial calcium uptake may serve as novel biomarkers for predicting poor prognosis in HCC. This study includes tumor specimens and adjacent normal liver tissue from 354 patients with confirmed HCC as study subjects and concluded that HCC patients with low MICU1 and high MCU/MICU2 expression exhibited poor survival rates, overall survival rates and disease-free survival rates [87]. Another study shows that the MCUR1 promotes in vitro invasion and in vivo metastasis of HCC cells by promoting epithelial−mesenchymal transition. This process is mainly done by the MCUR1 through the activation of the ROS/Nrf2/Notch1 pathway. It has also been found that treatment with the mitochondrial Ca2+-buffering protein parvalbumin significantly inhibits the ROS/Nrf2/Notch pathway, MCUR1-induced epithelial− mesenchymal transition and HCC metastasis [88]. In a study of CRC, the MCU is markedly increased in CRC tissues, and upregulated MCU is associated with poor prognosis in patients with CRC [89]. An upregulated MCU enhances mitochondrial Ca2+ uptake and causes mitochondrial Ca2+ imbalance, which, in turn, promotes CRC cell growth in vitro and in vivo. Ru360 is a highly potent and selective MCU inhibitor that can effectively block MCU-mediated mitochondrial Ca2+ uptake and, ultimately, slow CRC progress. These results may provide a potential pharmacological target for CRC treatment [90]. Saverio Marchi’s group demonstrated for the first time that MCUs are suitable targets for miRNA-25, which reduces prostate and colon cancer cells’ dependence on Ca2+ [91]. Therefore, the apoptosis could be induced in cancer cells by reducing MCU protein levels and mitochondrial Ca2+ uptake.
References
- Denton, R. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 2009, 1787, 1309–1316.
- Schönekess, B.O.; Brindley, P.G.; Lopaschuk, G.D. Calcium regulation of glycolysis, glucose oxidation, and fatty acid oxidation in the aerobic and ischemic heart. Can. J. Physiol. Pharmacol. 1995, 73, 1632–1640.
- McMillin, J.B.; Pauly, D.F. Control of mitochondrial respiration in muscle. Mol. Cell. Biochem. 1988, 81, 121–129.
- Konji, V.; Montag, A.; Sandri, G.; Nordenbrand, K.; Ernster, L. Transport of Ca2+ and Mn2+ by mitochondria from rat liver, heart and brain. Biochimie 1985, 67, 1241–1250.
- Oliveira, G.L.; Coelho, A.R.; Marques, R.; Oliveira, P.J. Cancer cell metabolism: Rewiring the mitochondrial hub. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166016.
- Iqbal, M.A.; Gupta, V.; Gopinath, P.; Mazurek, S.; Bamezai, R.N. Pyruvate kinase M2 and cancer: An updated assessment. FEBS Lett. 2014, 588, 2685–2692.
- Li, T.; Han, J.; Jia, L.; Hu, X.; Chen, L.; Wang, Y. PKM2 coordinates glycolysis with mitochondrial fusion and oxidative phosphorylation. Protein Cell. 2019, 10, 583–594.
- Mazurek, S. Pyruvate kinase type M2: A key regulator of the metabolic budget system in tumor cells. Int. J. Biochem. Cell Biol. 2011, 43, 969–980.
- Dey, P.; Kimmelman, A.C.; DePinho, R.A. Metabolic Codependencies in the Tumor Microenvironment. Cancer Discov. 2021, 11, 1067–1081.
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309.
- Ferro, F.; Servais, S.; Besson, P.; Roger, S.; Dumas, J.F.; Brisson, L. Autophagy and mitophagy in cancer metabolic remodelling. Semin. Cell Dev. Biol. 2020, 98, 129–138.
- Kim, K.H.; Lee, M.S. Autophagy--a key player in cellular and body metabolism. Nat. Rev. Endocrinol. 2014, 10, 322–337.
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San, P.J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863.
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473.
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12.
- Doherty, J.; Baehrecke, E.H. Life, death and autophagy. Nat. Cell Biol. 2018, 20, 1110–1117.
- White, E.; Mehnert, J.M.; Chan, C.S. Autophagy, Metabolism, and Cancer. Clin. Cancer Res. 2015, 21, 5037–5046.
- Hu, Y.X.; Han, X.S.; Jing, Q. Ca(2+) Ion and Autophagy. Adv. Exp. Med. Biol. 2019, 1206, 151–166.
- Bootman, M.D.; Chehab, T.; Bultynck, G.; Parys, J.B.; Rietdorf, K. The regulation of autophagy by calcium signals: Do we have a consensus? Cell Calcium. 2018, 70, 32–46.
- Lam, A.K.; Galione, A. The endoplasmic reticulum and junctional membrane communication during calcium signaling. Biochim. Biophys Acta 2013, 1833, 2542–2559.
- Høyer-Hansen, M.; Bastholm, L.; Szyniarowski, P.; Campanella, M.; Szabadkai, G.; Farkas, T.; Bianchi, K.; Fehrenbacher, N.; Elling, F.; Rizzuto, R.; et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol. Cell. 2007, 25, 193–205.
- Wang, Y.; Zhang, J.; Jiang, P.; Li, K.; Sun, Y.; Huang, Y. ASIC1a promotes acidic microenvironment-induced HCC cells migration and invasion by inducing autophagy. Eur. J. Pharmacol. 2021, 907, 174252.
- Li, M.; Kondo, T.; Zhao, Q.L.; Li, F.J.; Tanabe, K.; Arai, Y.; Zhou, Z.C.; Kasuya, M. Apoptosis induced by cadmium in human lymphoma U937 cells through Ca2+-calpain and caspase-mitochondria- dependent pathways. J. Biol. Chem. 2000, 275, 39702–39709.
- Cárdenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgó, J.; Müller, M.; Vais, H.; Cheung, K.H.; Yang, J.; Parker, I.; et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010, 142, 270–283.
- Decuypere, J.P.; Paudel, R.C.; Parys, J.; Bultynck, G. Intracellular Ca(2+) signaling: A novel player in the canonical mTOR-controlled autophagy pathway. Commun. Integr. Biol. 2013, 6, e25429.
- Bidaux, G.; Gordienko, D.; Shapovalov, G.; Farfariello, V.; Borowiec, A.; Iamshanova, O.; Lemonnier, L.; Gueguinou, M.; Guibon, R.; Fromont, G.; et al. 4TM-TRPM8 channels are new gatekeepers of the ER-mitochondria Ca2+ transfer. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 981–994.
- Cardenas, C.; Lovy, A.; Silva-Pavez, E.; Urra, F.; Mizzoni, C.; Ahumada-Castro, U.; Bustos, G.; Jaňa, F.; Cruz, P.; Farias, P.; et al. Cancer cells with defective oxidative phosphorylation require endoplasmic reticulum-to-mitochondria Ca2+ transfer for survival. Sci. Signal. 2020, 13, eaay1212.
- Kiviluoto, S.; Schneider, L.; Luyten, T.; Vervliet, T.; Missiaen, L.; De Smedt, H.; Parys, J.B.; Methner, A.; Bultynck, G. Bax inhibitor-1 is a novel IP3 receptor-interacting and -sensitizing protein. Cell Death Dis. 2012, 3, e367.
- Doghman-Bouguerra, M.; Lalli, E. ER-mitochondria interactions: Both strength and weakness within cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 650–662.
- Vultur, A.; Gibhardt, C.S.; Stanisz, H.; Bogeski, I. The role of the mitochondrial calcium uniporter (MCU) complex in cancer. Pflugers Arch. 2018, 470, 1149–1163.
- Yu, C.; Wang, Y.; Peng, J.; Shen, Q.; Chen, M.; Tang, W.; Li, X.; Cai, C.; Wang, B.; Cai, S.; et al. Mitochondrial calcium uniporter as a target of microRNA-340 and promoter of metastasis via enhancing the Warburg effect. Oncotarget 2017, 8, 83831–83844.
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516.
- Fadeel, B.; Zhivotovsky, B.; Orrenius, S. All along the watchtower: On the regulation of apoptosis regulators. FASEB J. 1999, 13, 1647–1657.
- Allan, L.; Clarke, P. Apoptosis and autophagy: Regulation of caspase-9 by phosphorylation. FEBS J. 2009, 276, 6063–6073.
- Abate, M.; Festa, A.; Falco, M.; Lombardi, A.; Luce, A.; Grimaldi, A.; Zappavigna, S.; Sperlongano, P.; Irace, C.; Caraglia, M.; et al. Mitochondria as playmakers of apoptosis, autophagy and senescence. Semin. Cell Dev. Biol. 2020, 98, 139–153.
- Sinha, K.; Das, J.; Pal, P.B.; Sil, P.C. Oxidative stress: The mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch. Toxicol. 2013, 87, 1157–1180.
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100.
- Jeong, S.; Seol, D. The role of mitochondria in apoptosis. BMB Rep. 2008, 41, 11–22.
- Chen, X.; Zhang, X.; Kubo, H.; Harris, D.M.; Mills, G.D.; Moyer, J.; Berretta, R.; Potts, S.T.; Marsh, J.D.; Houser, S.R. Ca2+ influx-induced sarcoplasmic reticulum Ca2+ overload causes mitochondrial-dependent apoptosis in ventricular myocytes. Circ. Res. 2005, 97, 1009–1017.
- McConkey, D.J. Biochemical determinants of apoptosis and necrosis. Toxicol. Lett. 1998, 99, 157–168.
- Aharoni-Simon, M.; Shumiatcher, R.; Yeung, A.; Shih, A.Z.; Dolinsky, V.W.; Doucette, C.A.; Luciani, D.S. Bcl-2 Regulates Reactive Oxygen Species Signaling and a Redox-Sensitive Mitochondrial Proton Leak in Mouse Pancreatic β-Cells. Endocrinology 2016, 157, 2270–2281.
- Warren, C.F.A.; Wong-Brown, M.W.; Bowden, N.A. BCL-2 family isoforms in apoptosis and cancer. Cell Death Dis. 2019, 10, 177.
- Selzer, E.; Schlagbauer-Wadl, H.; Okamoto, I.; Pehamberger, H.; Pötter, R.; Jansen, B. Expression of Bcl-2 family members in human melanocytes, in melanoma metastases and in melanoma cell lines. Melanoma Res. 1998, 8, 197–203.
- Berridge, M.J. The endoplasmic reticulum: A multifunctional signaling organelle. Cell Calcium. 2002, 32, 235–249.
- Berridge, M.J. The Inositol Trisphosphate/Calcium Signaling Pathway in Health and Disease. Physiol. Rev. 2016, 96, 1261–1296.
- Kondratskyi, A.; Kondratska, K.; Skryma, R.; Prevarskaya, N. Ion channels in the regulation of apoptosis. Biochim. Biophys. Acta 2015, 1848, 2532–2546.
- Nakagawa, T.; Zhu, H.; Morishima, N.; Li, E.; Xu, J.; Yankner, B.A.; Yuan, J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 2000, 403, 98–103.
- Feissner, R.F.; Skalska, J.; Gaum, W.E.; Sheu, S.S. Crosstalk signaling between mitochondrial Ca2+ and ROS. Front. Biosci. 2009, 14, 1197–1218.
- Yan, X.; Xun, M.; Li, J.; Wu, L.; Dou, X.; Zheng, J. Activation of Na+/K+-ATPase attenuates high glucose-induced H9c2 cell apoptosis via suppressing ROS accumulation and MAPKs activities by DRm217. Acta. Biochim. Biophys. Sin. 2016, 48, 883–893.
- Madreiter-Sokolowski, C.T.; Thomas, C.; Ristow, M. Interrelation between ROS and Ca2+ in aging and age-related diseases. Redox. Biol. 2020, 36, 101678.
- Raimondi, M.; Fontana, F.; Marzagalli, M.; Audano, M.; Beretta, G.; Procacci, P.; Sartori, P.; Mitro, N.; Limonta, P. Ca2+ overload- and ROS-associated mitochondrial dysfunction contributes to δ-tocotrienol-mediated paraptosis in melanoma cells. Apoptosis 2021, 26, 277–292.
- Ataizi, Z.S.; Ertilav, K.; Nazıroğlu, M. Mitochondrial oxidative stress-induced brain and hippocampus apoptosis decrease through modulation of caspase activity, Ca2+ influx and inflammatory cytokine molecular pathways in the docetaxel-treated mice by melatonin and selenium treatments. Metab. Brain Dis. 2019, 34, 1077–1089.
- Li, W.; Liu, B.; Wang, L.; Liu, J.; Yang, X.; Zheng, J. Melatonin Attenuates Cardiac Ischemia-Reperfusion Injury through Modulation of IP3R-Mediated Mitochondria-ER Contact. Oxid. Med. Cell. Longev. 2021, 1370862.
- Meis, L. Role of the sarcoplasmic reticulum Ca2+-ATPase on heat production and thermogenesis. Biosci. Rep. 2001, 21, 113–137.
- Zhou, D.; Shao, L.; Spitz, D. Reactive oxygen species in normal and tumor stem cells. Adv. Cancer Res. 2014, 122, 1–67.
- Hsu, C.C.; Tseng, L.M.; Lee, H.C. Role of mitochondrial dysfunction in cancer progression. Exp. Biol. Med. 2016, 241, 1281–1295.
- Shoshan-Barmatz, V.; Krelin, Y.; Shteinfer-Kuzmine, A. VDAC1 functions in Ca2+ homeostasis and cell life and death in health and disease. Cell Calcium. 2018, 69, 81–100.
- Szatrowski, T.; Nathan, C. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798.
- Tsunoda, T.; Koga, H.; Yokomizo, A.; Tatsugami, K.; Eto, M.; Inokuchi, J.; Hirata, A.; Masuda, K.; Okumura, K.; Naito, S. Inositol 1,4,5-trisphosphate (IP3) receptor type1 (IP3R1) modulates the acquisition of cisplatin resistance in bladder cancer cell lines. Oncogene 2005, 24, 1396–1402.
- Brouland, J.P.; Valleur, P.; Papp, B. Expression of SERCA pumps during cell differentiation and tumorigenesis: Application to colonic carcinogenesis. Ann. Pathol. 2006, 26, 159–172.
- Kuchay, S.; Giorgi, C.; Simoneschi, D.; Pagan, J.; Missiroli, S.; Saraf, A.; Florens, L.; Washburn, M.P.; Collazo-Lorduy, A.; Castillo-Martin, M.; et al. PTEN counteracts FBXL2 to promote IP3R3- and Ca2+-mediated apoptosis limiting tumour growth. Nature 2017, 546, 554–558.
- Bhagat, S.; Singh, S. Co-delivery of AKT3 siRNA and PTEN Plasmid by Antioxidant Nanoliposomes for Enhanced Antiproliferation of Prostate Cancer Cells. ACS Appl. Bio. Mater. 2020, 3, 3999–4011.
- Bononi, A.; Giorgi, C.; Patergnani, S.; Larson, D.; Verbruggen, K.; Tanji, M.; Pellegrini, L.; Signorato, V.; Olivetto, F.; Pastorino, S.; et al. BAP1 regulates IP3R3-mediated Ca2+ flux to mitochondria suppressing cell transformation. Nature 2017, 546, 549–553.
- Jin, C.; Kumar, P.; Gracia-Sancho, J.; Dufour, J.F. Calcium transfer between endoplasmic reticulum and mitochondria in liver diseases. FEBS Lett. 2021, 595, 1411–1421.
- Boutin, B.; Tajeddine, N.; Monaco, G.; Molgo, J.; Vertommen, D.; Rider, M.; Parys, J.B.; Bultynck, G.; Gailly, P. Endoplasmic reticulum Ca(2+) content decrease by PKA-dependent hyperphosphorylation of type 1 IP3 receptor contributes to prostate cancer cell resistance to androgen deprivation. Cell Calcium. 2015, 57, 312–320.
- Cui, C.; Merritt, R.; Fu, L.; Pan, Z. Targeting calcium signaling in cancer therapy. Acta Pharm. Sin. B 2017, 7, 3–17.
- Romero-Garcia, S.; Prado-Garcia, H. Mitochondrial calcium: Transport and modulation of cellular processes in homeostasis and cancer (Review). Int. J. Oncol. 2019, 54, 1155–1167.
- Colwill, K.; Gräslund, S. A roadmap to generate renewable protein binders to the human proteome. Nat. Methods 2011, 8, 551–558.
- Zeng, F.; Chen, X.; Cui, W.; Wen, W.; Lu, F.; Sun, X.; Ma, D.; Yuan, Y.; Li, Z.; Hou, N.; et al. RIPK1 Binds MCU to Mediate Induction of Mitochondrial Ca2+ Uptake and Promotes Colorectal Oncogenesis. Cancer Res. 2018, 78, 2876–2885.
- Wu, R.; Zuo, W.; Xu, X.; Bi, L.; Zhang, C.; Chen, H.; Liu, H. MCU That Is Transcriptionally Regulated by Nrf2 Augments Malignant Biological Behaviors in Oral Squamous Cell Carcinoma Cells. BioMed Res. Int. 2021, 6650791.
- Zheng, S.; Wu, R.; Deng, Y.; Zhang, Q. Dihydroartemisinin represses oral squamous cell carcinoma progression through downregulating mitochondrial calcium uniporter. Bioengineered 2022, 13, 227–241.
- Marchi, S.; Vitto, V.A.M.; Patergnani, S.; Pinton, P. High mitochondrial Ca2+ content increases cancer cell proliferation upon inhibition of mitochondrial permeability transition pore (mPTP). Cell Cycle 2019, 18, 914–916.
- Patergnani, S.; Guzzo, S.; Mangolini, A.; dell’Atti, L.; Pinton, P.; Aguiari, G. The induction of AMPK-dependent autophagy leads to P53 degradation and affects cell growth and migration in kidney cancer cells. Exp. Cell Res. 2020, 395, 112190.
- Fei, M.; Zhang, L.; Wang, H.; Zhu, Y.; Niu, W.; Tang, T.; Han, Y. Inhibition of Cathepsin S Induces Mitochondrial Apoptosis in Glioblastoma Cell Lines Through Mitochondrial Stress and Autophagosome Accumulation. Front. Oncol. 2020, 10, 516746.
- Xue, P.; Chen, Q.; Ren, X.; Liu, D.; Yang, X. A novel protoapigenone analog RY10-4 induces apoptosis of breast cancer cells by exacerbating mitochondrial Ca2+ influx through mitochondrial calcium uniporter. Toxicol. Appl. Pharmacol. 2021, 433, 115776.
- Ren, T.; Wang, J.; Zhang, H.; Yuan, P.; Zhu, J.; Wu, Y.; Huang, Q.; Guo, X.; Zhang, J.; Ji, L.; et al. MCUR1-Mediated Mitochondrial Calcium Signaling Facilitates Cell Survival of Hepatocellular Carcinoma via Reactive Oxygen Species-Dependent P53 Degradation. Antioxid. Redox Signal. 2018, 28, 1120–1136.
- Marchi, S.; Pinton, P. Mitochondrial calcium uniporter, MiRNA and cancer: Live and let die. Commun. Integr. Biol. 2013, 6, e23818.
- Sun, Y.; Li, M.; Liu, G.; Zhang, X.; Zhi, L.; Zhao, J.; Wang, G. The function of Piezo1 in colon cancer metastasis and its potential regulatory mechanism. J. Cancer Res. Clin. Oncol. 2020, 146, 1139–1152.
- Wang, X.; Li, Y.; Li, Z.; Lin, S.; Wang, H.; Sun, J.; Lan, C.; Wu, L.; Sun, D.; Huang, C.; et al. Mitochondrial calcium uniporter drives metastasis and confers a targetable cystine dependency in pancreatic cancer. Cancer Res. 2022, 3230, 2021.
- Tosatto, A.; Sommaggio, R.; Kummerow, C.; Bentham, R.B.; Blacker, T.S.; Berecz, T.; Duchen, M.R.; Rosato, A.; Bogeski, I.; Szabadkai, G.; et al. The mitochondrial calcium uniporter regulates breast cancer progression via HIF-1α. EMBO Mol. Med. 2016, 8, 569–585.
- Tang, S.; Wang, X.; Shen, Q.; Yang, X.; Yu, C.; Cai, C.; Cai, G.; Meng, X.; Zou, F. Mitochondrial Ca2+ uniporter is critical for store-operated Ca2+ entry-dependent breast cancer cell migration. Biochem. Biophys. Res. Commun. 2015, 458, 186–193.
- Gao, P.; Peng, T.; Lin, S.; Zhi, W.; Cao, C.; Wu, P.; Xi, L.; Wu, P.; Yang, Q.; Ding, W. Key Role of MCUR1 in Malignant Progression of Breast Cancer. OncoTargets Ther. 2021, 14, 4163–4175.
- Che, X.; Wang, Q.; Zhang, B. MCUR1 is a marker for the progression and prognosis of breast cancer. Res. Square 2022. preprint (Version 1).
- Ren, T.; Zhang, H.; Wang, J.; Zhu, J.; Jin, M.; Wu, Y.; Guo, X.; Ji, L.; Huang, Q.; Zhang, H.; et al. MCU-dependent mitochondrial Ca2+ inhibits NAD+/SIRT3/SOD2 pathway to promote ROS production and metastasis of HCC cells. Oncogene 2017, 36, 5897–5909.
- Bartha, A.; Győrffy, B. TNMplot.com: A Web Tool for the Comparison of Gene Expression in Normal, Tumor and Metastatic Tissues. Int. J. Mol. Sci. 2021, 22, 2622.
- Parekh, A. Calcium signalling in health and disease. Semin Cell Dev Biol. 2019, 94, 1–2.
- Li, C.; Lin, H.; Ko, C.; Lai, J.; Chu, P. A Novel Biomarker Driving Poor-Prognosis Liver Cancer: Overexpression of the Mitochondrial Calcium Gatekeepers. Biomedicines 2020, 8, 451.
- Jin, M.; Wang, J.; Ji, X.; Cao, H.; Zhu, J.; Chen, Y.; Yang, J.; Zhao, Z.; Ren, T.; Xing, J. MCUR1 facilitates epithelial-mesenchymal transition and metastasis via the mitochondrial calcium dependent ROS/Nrf2/Notch pathway in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 136.
- Zhao, Y.; Wang, Y.; Zhao, J.; Zhang, Z.; Jin, M.; Zhou, F.; Jin, C.; Zhang, J.; Xing, J.; Wang, N.; et al. PDE2 Inhibits PKA-Mediated Phosphorylation of TFAM to Promote Mitochondrial Ca2+-Induced Colorectal Cancer Growth. Front. Oncol. 2021, 11, 663778.
- Liu, Y.; Jin, M.; Wang, Y.; Zhu, J.; Tan, R.; Zhao, J.; Ji, X.; Jin, C.; Jia, Y.; Ren, T.; et al. MCU-induced mitochondrial calcium uptake promotes mitochondrial biogenesis and colorectal cancer growth. Signal Transduct. Target Ther. 2020, 5, 59.
- Marchi, S.; Vitto, V.A.M.; Danese, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial calcium uniporter complex modulation in cancerogenesis. Cell Cycle 2019, 18, 1068–1083.
More
Information
Subjects:
Endocrinology & Metabolism
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
953
Revisions:
2 times
(View History)
Update Date:
30 Jun 2022
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No