Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Maria Brito | -- | 1502 | 2022-06-21 18:26:55 | | | |
| 2 | Dean Liu | Meta information modification | 1502 | 2022-06-22 02:57:24 | | | | |
| 3 | Dean Liu | Meta information modification | 1502 | 2022-09-09 03:18:06 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Afonso, M.; Brito, M.A. Tumor Cell Signaling Pathways. Encyclopedia. Available online: https://encyclopedia.pub/entry/24295 (accessed on 26 July 2026).
Afonso M, Brito MA. Tumor Cell Signaling Pathways. Encyclopedia. Available at: https://encyclopedia.pub/entry/24295. Accessed July 26, 2026.
Afonso, Mariana, Maria Alexandra Brito. "Tumor Cell Signaling Pathways" Encyclopedia, https://encyclopedia.pub/entry/24295 (accessed July 26, 2026).
Afonso, M., & Brito, M.A. (2022, June 21). Tumor Cell Signaling Pathways. In Encyclopedia. https://encyclopedia.pub/entry/24295
Afonso, Mariana and Maria Alexandra Brito. "Tumor Cell Signaling Pathways." Encyclopedia. Web. 21 June, 2022.
Copy Citation
Increasing the understanding of carcinogenesis has allowed the delineation of crucial signaling pathways, which have shown essential roles in the regulation of stem cell functions
neuro-oncology
tyrosine kinase receptors
malignant gliomas
signaling pathways
carcinogenesis
integrins
histones
ubiquitin-proteasome system
gene fusions
transmembrane monocarboxylate transporters
1. Introduction
Increasing the understanding of carcinogenesis has allowed the delineation of crucial signaling pathways, which have shown essential roles in the regulation of stem cell functions [1][2]. Accurate and appropriate regulation is doubtless critical for biological activity. However, many of these pathways are dysregulated in cancer, becoming part of oncogenic transformation [2][3]. These transformations involve amplification or overexpression of oncogenes, along with loss or lack of expression of tumor suppressor genes [4]. Therefore, to perceive the logic behind the therapeutic options in gliomas, it is pertinent to understand the different signaling pathways and the transformations in glioma cells outlined in Figure 1 and detailed below.
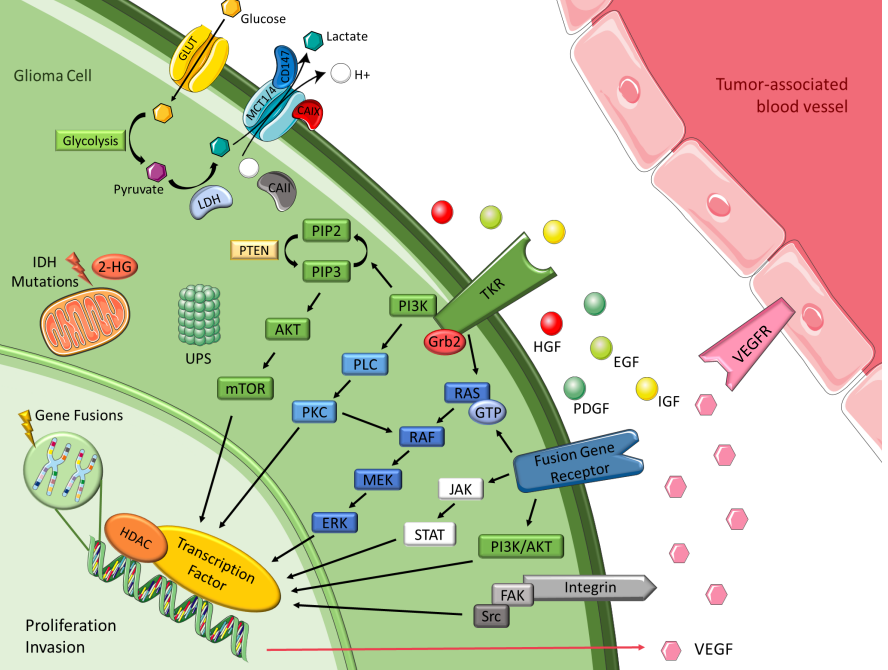
Figure 1. Malignant glioma signaling pathways. Figure drawn with smart.servier.com.
2. Tyrosine Kinase Receptor Pathways
Brain tumors cells oversecrete growth factors and overexpress their receptors, creating paracrine and autocrine stimulatory loops, wherein tyrosine kinase is the leading actor [4][5]. These growth factors include epidermal growth factor (EGF), platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF), and insulin-like growth factor (IGF). They are correlated with tyrosine kinase and share common mechanisms and intracellular signaling activation [4][6]. EGFR gene amplification is the most frequent alteration in GBM, and leads to EGFR protein overexpression, EGFRvIII being the most common mutation [5][6]. This mutation serves as a strong tumor-restricted antigen, as it is expressed in 30-40% of human GBM tumors to enhance glioma cell proliferation, angiogenesis and invasiveness, but is not expressed in healthy tissue [7]. As previously explained, malignant gliomas need extensive vascularity, so they commonly feature high expression of VEGF to allow new blood vessel formation, whereas tumor-associated endothelial cells express the corresponding receptor, vascular endothelial growth factor receptor 2 (VEGFR-2) [6]. Moreover, platelet derived growth factor receptor (PDGFR) overexpression has been observed in gliomas and associated with tumor growth and angiogenesis, leading to malignant progression [5][6]. GBM cell lines exhibit upregulation of insulin-like growth factor-1 receptor (IGF-1R), resulting in proliferative, antiapoptotic and proinvasive effects [8].
3. Intracellular Downstream Pathways
Most intracellular effectors are serine or threonine kinases, such as rat sarcoma (Ras), phosphoinositide 3-kinase (PI3K) or phospholipase C (PLC), which are recruited to the cell membrane after activation of tyrosine kinase receptor (TKR) and rely on adaptor proteins such as growth factor receptor-bound protein 2 (Grb2) to relay the signals from the cell membrane [6][9]. Gliomas are correlated with either activation of these effector molecules or inactivating mutations of the negative regulators of these kinases, leading in both cases to accentuated effects and dysregulation [6]. Gliomas barely express oncogenic Ras mutations, but they often have increased Ras activity due to a mutation or amplification of upstream positive regulators, such as EGFR and PDGFR [5][6]. The mitogen-activated protein kinase (MAPK) pathway is initiated due to the farnesylation of Ras that is then catalyzed by farnesyltransferase enzyme to activate the rapidly accelerated fibrosarcoma (RAF), mitogen-activated protein extracellular regulated kinase (MEK), extracellular signal-regulated kinases (ERK) to induce the translocation of proteins to the nucleus in order to promote cell cycle progression and anti-apoptosis genes [5][10]. The phosphatidylinositol (3,4,5)-trisphosphate (PIP3)/ protein kinase B (AKT)/ mechanistic target of rapamycin (mTOR) pathway involves TKR and the loss of phosphatase and tensin homolog (PTEN), a negative regulator of these kinases, culminating in deregulation of proliferation, growth, apoptosis, and cytoskeletal rearrangement [5][6]. Protein kinase C (PKC) regulates cell proliferation, invasion and angiogenesis [6]. It is located at the crossroads of multiple signaling pathways and acts like a relay station for signals to the nucleus. PKC is activated by phospholipase-C (PLC), as well as by PI3K, and then transmits signal to the nucleus via the MAPK pathway (mainly) and via the PI3K/AKT pathway [8].
4. Histones
Histones are basic proteins that order and package DNA into nucleosomes, playing a crucial role in the regulation of gene expression. Histone deacetylases (HDAC) are enzymes responsible for catalyzing the removal of acetyl functional groups from the lysine groups of both histone and nonhistone proteins. Changes in histones affect transcription, repair, and replication, leading to alterations in cell proliferation, survival and differentiation. This is the reason why they have been considered a key target for cancer therapeutics. Some studies have revealed that both genetic and epigenetic mechanisms are significantly deregulated in glioma cells. In particular, modifications in sequence or expression of gene coding for HDACs may contribute to GBM pathogenesis and progression [11][12][13].
5. Integrins
Integrins represent a family of transmembrane adhesion receptors that integrate signals between cells and the surrounding stroma. They lack intrinsic catalytic activity, implying the presence of external signals for them to be effective. Their effectiveness occurs throughout the activation of integrin-associated proteins and binding of focal adhesions with non-receptor tyrosine kinases, such as focal adhesion kinase (FAK) and Src. Once integrin dimers are formed, downstream signaling pathways are activated, regulating migration, invasion, angiogenesis, and survival. Integrin-encoding genes are rarely mutated in cancers, but deregulation of integrin signaling is quite frequent. Glioma cells invade brain parenchyma preferably along higher rigidity tracts, such as the vascular basement membrane that contains various integrin ligands [14][15].
6. Ubiquitin-Proteasome System
The ubiquitin-proteasome system (UPS) is a complex and universal protein degradation pathway essential to ensure the balance between cell proliferation and apoptosis [16]. High grade tumors have an inherent potential of escaping cell cycle control mechanisms such as UPS, which results in uncontrolled cell division [8][14]. Hence the importance of these mechanisms being in constant balance and making this a great therapeutic target.
7. Gene Fusions
Gene fusions are hybrid genes constituted by the combination of the DNA sequences of two genes known to be oncogenic drivers in multiple malignancies. They have the potential to form chimeric proteins with altered functions, and remain an active area of research [17][18]. The Janus kinase (JAK)-STAT pathway initiates transcription of regions of the genome, inducing the expression of anti-apoptotic proteins and other cell cycle regulators, which leads to cellular growth and proliferation [19]. The incidence of gene fusions has been increasingly recognized in GBM, occurring in up to 50% of tumors, with targetable fusions involving a tyrosine kinase domain in approximately 10%. These predominantly include neurotrophic tyrosine receptor kinase (NTRK), fibroblast growth factor receptor (FGFR) and mesenchymal-epithelial transition factor (MET) fusions [20]. NTRK are encoded by three different genes, NTRK1, NTRK2 and NTRK3. Genomic rearrangements in NTRK genes result in gene fusions and trigger activation of oncogenic TRK signaling [14]. FGFR is the most common fusion expressed in GBM, specifically fibroblast growth factor receptor 3–transforming acidic coiled coil-containing protein 3 (FGFR3:TACC3), which is the fusion with relevance as a potential target in multiple cancers, including GBM [14]. MET encodes the receptor for hepatocyte growth factor (HGF) and has great importance in the migration and invasiveness of glioma cells, such as in response to irradiation, inhibition of angiogenesis and hypoxia, and has a critical role in therapeutic resistance and recurrence of GBM [9][14][21].
8. Isocitrate Dehydrogenase
IDH enzymes play essential roles in the Krebs cycle and cellular homeostasis by catalyzing the oxidative decarboxylation of isocitrate. There are three isoforms, with IDH1 in the cytoplasm and peroxisomes and IDH2 and IDH3 in the mitochondrial matrix. Advances in cancer genetics revealed that the genes encoding IDHs are prevalent in human malignancies, including gliomas. One of the consequences of IDH mutation is the alteration of enzymatic activity, which leads to the synthesis of 2-hydroxyglutarate (2-HG) and has been implicated in epigenetic mechanisms of oncogenesis. 2-HG elevation suppresses the electron transport chain, interfering with cellular metabolism and epigenetic regulation. However, the tumor-initiating and progressing capacity of 2-HG had not been fully demonstrated [22][23][24][25].
9. Transmembrane Monocarboxylate Transporters
A common phenomenon that characterizes the adaptation of tumor cells is the shift from oxidative phosphorylation to aerobic glycolysis as a main source of ATP. A high rate of glycolysis leads to the overproduction of lactic acid, which is associated with poor prognosis. Glucose enters the cell through the glucose transporter and is converted into pyruvate, which turns into lactate due to lactate dehydrogenase activity [26]. Monocarboxylate metabolites such as l-lactate are transported by the monocarboxylate transporters (MCTs) family members MCT1, MCT2, MCT3 and MCT4 [27]. Therefore, these transporters allow glycolysis to operate at a high speed by mediating the efflux of lactic acid into the extracellular environment, which also helps preventing acidosis, thus playing an important role in pH regulation [27]. Among these MCTs, MCT1 and MCT4 are present in astrocytes and stand out in glioma cells, playing a pivotal role in tumor cell survival by promoting lactate efflux [26][28]. MCT2 is the primary isoform expressed in human GBM and glioma-derived cell lines and is expressed in the neurons [29], which use the lactate produced by astrocytes. In the brain, glycolytic oligodendrocytes and astrocytes export lactate through MCT1 and MCT4 to fuel oxidative neurons expressing MCT2 [27]. It was hypothesized that carbonic anhydrases (CAII/CAIX) functions as a proton-collecting antenna, thereby enhancing the activity of proton-coupled MCTs, while CD147 acts as a chaperone facilitating membrane trafficking of MCT1/4 [30][31]. Accordingly, the plasma membrane localization of MCT1 and MCT4 was shown to be regulated by coexpression with the chaperone protein CD147, which was also upregulated in GBM compared to diffuse astrocytomas and nonneoplastic brain [26].
References
- Weller, M. Next generation neuro-oncology. Eur. J. Cancer 2018, 96, 1–5.
- Matsui, W.H. Cancer stem cell signaling pathways. Medicine 2016, 95, S8–S19.
- Clark, P.A.; Treisman, D.M.; Ebben, J.; Kuo, J.S. Developmental Signaling Pathways in Brain Tumor-Derived Stem-Like Cells. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2007, 236, 3297–3308.
- Newton, H.B. Molecular neuro-oncology and development of targeted therapeutic strategies for brain tumors. Part 1: Growth factor and Ras signaling pathways. Expert Rev. Anticancer. Ther. 2003, 3, 595–614.
- Nakada, M.; Kita, D.; Watanabe, T.; Hayashi, Y.; Teng, L.; Pyko, I.V.; Hamada, J. Aberrant Signaling Pathways in Glioma. Cancers 2011, 3, 3242–3278.
- Sathornsumetee, S.; Desjardins, A.; Quinn, J.A.; Vredenburgh, J.J.; Rich, J.N. Molecularly Targeted Therapy for Malignant Glioma. Cancer 2007, 110, 13–24.
- Shahpar, S.; Mhatre, P.V.; Huang, M.E. Update on Brain Tumors: New Developments in Neuro-oncologic Diagnosis and Treatment, and Impact on Rehabilitation Strategies. PMR 2016, 8, 678–689.
- Sathornsumetee, S.; Rich, J.N. Molecularly Targeted Therapy in Neuro-Oncology. In Handbook of Clinical Neurology, 1st ed.; Aminoff, M.J., Boller, F., Swaab, D.F., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; Volume 104, pp. 255–278. ISBN 1216636079.
- Tilak, M.; Holborn, J.; New, L.A.; Lalonde, J.; Jones, N. Receptor Tyrosine Kinase Signaling and Targeting in Glioblastoma Multiforme. Int. J. Mol. Sci. 2021, 22, 1831.
- Harrison, R.A.; Groot, J.F. De Cell Signaling Pathways in Brain Tumors. Top. Magn. Reson. Imaging 2017, 26, 15–26.
- Was, H.; Krol, S.K.; Rotili, D.; Mai, A.; Wojtas, B.; Kaminska, B.; Maleszewska, M. Histone deacetylase inhibitors exert anti-tumor effects on human adherent and stem-like glioma cells. Clin. Epigenet. 2019, 11, 11.
- Chessum, N.; Jones, K.; Pasqua, E.; Tucker, M. Recent Advances in Cancer Therapeutics. In Progress in Medicinal Chemistry, 1st ed.; Lawton, G., Witty, D.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; Volume 54, pp. 1–63.
- Seto, E.; Yoshida, M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713.
- Le Rhun, E.; Preusser, M.; Roth, P.; Reardon, D.A.; van den Bent, M.; Wen, P.; Reifenberger, G.; Weller, M. Molecular targeted therapy of glioblastoma. Cancer Treat. Rev. 2019, 80, 101896.
- Ellert-miklaszewska, A.; Poleszak, K.; Pasierbinska, M. Integrin Signaling in Glioma Pathogenesis: From Biology to Therapy. Int. J. Mol. Sci. 2020, 21, 888.
- Vlachostergios, P.J.; Voutsadakis, I.A.; Papandreou, C.N. The ubiquitin-proteasome system in glioma cell cycle control. Cell Div. 2012, 7, 18.
- Young Woo, H.; Na, K.; Yoo, J.; Chang, J.H.; Park, Y.N.; Hyo, S.; Kim, S.H. Glioblastomas harboring gene fusions detected by next-generation sequencing. Brain Tumor Pathol. 2020, 37, 136–144.
- Ferguson, S.D.; Zhou, S.; Huse, J.T.; de Groot, J.F.; Xiu, J.; Subramaniam, D.S.; Mehta, S.; Gatalica, Z.; Swensen, J.; Sanai, N.; et al. Targetable gene fusions associate with the IDH wild-type astrocytic lineage in adult gliomas. J. Neuropathol. Exp. Neurol. 2018, 77, 437–442.
- You, G.; Fan, X.; Hu, H.; Jiang, T.; Chen, C.C. Fusion Genes Altered in Adult Malignant Gliomas. Front. Neurol. 2021, 12, 715206.
- Tan, A.C.; Ashley, D.M.; López, G.Y. Management of Glioblastoma: State of the Art and Future Directions. CA A Cancer J. Clin. 2020, 70, 299–312.
- Cheng, F.; Guo, D. MET in glioma: Signaling pathways and targeted therapies. J. Exp. Clin. Cancer Res. 2019, 38, 270.
- Han, S.; Liu, Y.; Cai, S.J.; Qian, M.; Ding, J.; Larion, M.; Gilbert, M.R.; Yang, C. IDH mutation in glioma: Molecular mechanisms and potential therapeutic targets. Br. J. Cancer 2020, 122, 1580–1589.
- Karpel-massler, G.; Nguyen, T.; Shang, E.; Siegelin, M.D.; Biology, C. Novel IDH1 Targeted Glioma Therapies. CNS Drugs. 2020, 33, 1155–1166.
- Chen, R.; Cohen, A.L.; Colman, H. Targeted Therapeutics in Patients With High-Grade Gliomas: Past, Present, and Future. Curr. Treat. Options Oncol. 2016, 17, 42.
- Touat, M.; Idbaih, A.; Sanson, M.; Ligon, K.L. Glioblastoma targeted therapy: Updated approaches from recent biological insights. Ann. Oncol. 2017, 28, 1457–1472.
- Miranda-Goncalves, V.; Honavar, M.; Pinheiro, C.; Martinho, O.; Pires, M.M.; Pinheiro, C.; Cordeiro, M.; Bebiano, G.; Costa, P.; Palmeirim, I.; et al. Monocarboxylate transporters (MCTs) in gliomas: Expression and exploitation as therapeutic targets. Neuro-Oncol. 2013, 15, 172–188.
- Pérez-escuredo, J.; Van Hée, V.F.; Sboarina, M.; Falces, J.; Payen, V.L.; Pellerin, L.; Sonveaux, P. Monocarboxylate transporters in the brain and in cancer. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2016, 1863, 2481–2497.
- Park, S.J.; Smith, C.P.; Wilbur, R.R.; Cain, C.P.; Kallu, S.R.; Valasapalli, S.; Sahoo, A.; Guda, M.R.; Tsung, A.J.; Velpula, K.K. An overview of MCT1 and MCT4 in GBM: Small molecule transporters with large implications. Am. J. Cancer Res. 2018, 8, 1967–1976.
- Khalyfa, A.; Qiao, Z.; Raju, M.; Shyu, C.R.; Coghill, L.; Ericsson, A.; Gozal, D. Monocarboxylate transporter-2 expression restricts tumor growth in a murine model of lung cancer: A multi-omic analysis. Int. J. Mol. Sci. 2021, 22, 10616.
- Felmlee, M.A.; Jones, R.S.; Rodriguez-cruz, V.; Follman, K.E.; Morris, M.E. Monocarboxylate Transporters (SLC16): Function, Regulation, and Role in Health and Disease. Pharmacol. Rev. 2020, 72, 466–485.
- Becker, H.M.; Deitmer, J.W. Proton Transport in Cancer Cells: The Role of Carbonic Anhydrases. Int. J. Mol. Sci. 2021, 22, 3171.
More
Information
Subjects:
Oncology; Medicine, Research & Experimental
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
3 times
(View History)
Update Date:
09 Sep 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No