Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Nazar Trotsko | -- | 3173 | 2022-06-17 18:42:27 | | | |
| 2 | Amina Yu | -29 word(s) | 3144 | 2022-06-20 04:01:36 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Trotsko, N.; Szczepański, J.; , . Anticancer Profile of Rhodanines. Encyclopedia. Available online: https://encyclopedia.pub/entry/24174 (accessed on 24 July 2026).
Trotsko N, Szczepański J, . Anticancer Profile of Rhodanines. Encyclopedia. Available at: https://encyclopedia.pub/entry/24174. Accessed July 24, 2026.
Trotsko, Nazar, Jacek Szczepański, . "Anticancer Profile of Rhodanines" Encyclopedia, https://encyclopedia.pub/entry/24174 (accessed July 24, 2026).
Trotsko, N., Szczepański, J., & , . (2022, June 17). Anticancer Profile of Rhodanines. In Encyclopedia. https://encyclopedia.pub/entry/24174
Trotsko, Nazar, et al. "Anticancer Profile of Rhodanines." Encyclopedia. Web. 17 June, 2022.
Copy Citation
The rhodanine derivatives are small compounds with a broad spectrum of biological activities; they are used as antimicrobial, antiviral, antitubercular, anti-inflammatory, antidiabetic, and antitumor agents. In the pharmaceutical market, epalrestat (rhodanine-3-acetic acid) has been marketed in Japan since 1992 for treatment of diabetic complications (peripheral neuropathy). Epalrestat is an inhibitor of aldose reductase, the key enzyme in the polyol pathway of glucose metabolism under hyperglycemic conditions. The good clinical safety profile of epalrestat justified the interest of the researchers in rhodanines as potential drug candidates.
rhodanines
anticancer activity
molecular targets
1. Introduction
Rhodanines were found to induce apoptosis through the modulation of the Bcl-2 family proteins [1][2] or through the modulation of other key signaling proteins [3][4]. Moreover, rhodanines were also reported to reveal their anticancer activity through the inhibition of the phosphatase of regenerating liver (PRL-3) [5].
Furthermore, 5-benzylidene-3-ethyl-rhodanine, also known as BRT-1, is an active anticancer agent which causes S-phase arrest and affects DNA replication in leukemic cells. BTR-1 activates apoptosis and induces cell death [6]. Some of these molecules could become effective and quite selective anticancer drugs in the future.
2. Rhodanines with Anticancer Properties
2.1. 3-Substituted Rhodanine Derivatives
Nguyen et al. synthesized a series of new structures, N-(4-oxo-2-thioxothiazolidin-3-yl)-2-[(4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)thio]acetamide derivatives, and evaluated them for their cytotoxicity potential against K562 (human chronic myelogenous leukemia) and MCF-7 (human breast adenocarcinoma) tumor cell lines. Compound 1 with the 2-thioxothiazolidin-4-one ring containing the active methylene group (Figure 1), as shown below, exerted moderate cytotoxicity against MCF-7 cells with a % inhibition of cell growth of 64.4% at the concentration of 100 µg/mL [7].
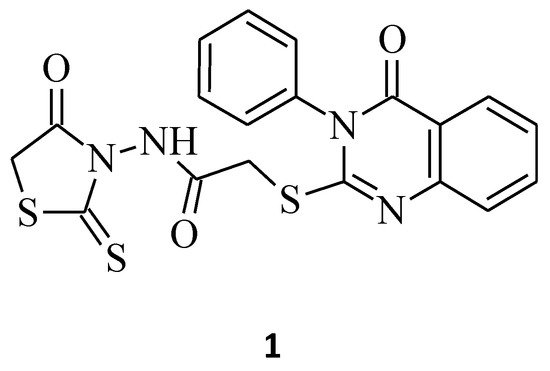
Figure 1. The structure of N-(4-oxo-2-thioxothiazolidin-3-yl)-2-[(4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)thio]acetamide.
On the other hand, the introduction of small groups such as -CH2COOH, -CH(CH3)COOH in the N-3 position of the rhodanine ring resulted in the formation of the N-substituted compounds 2 and 3, respectively (Figure 2). These molecules showed good antiproliferative activity in the human chronic myelogenous leukemia cell line K562, with an IC50 of 14.60, 11.10 µg/mL, respectively, and were twice or three times more potent than the other compounds from the study. Worth noticing is that these compounds were only 3- or 2.3-fold less active in comparison to the reference cisplatin (IC50 = 4.78 µg/mL) [8]. The introduction into structure 2 of the methyl group to carboxymethyl moiety only slightly increases the activity. It may have been caused by the similarity of the surface area of the N-3 substituent. However, further enlarging the methyl substituent to isopropyl, carboxyethyl, or benzyl substituents into position 3 of the rhodanine ring leads to a 2- or 3-fold decrease in activity.
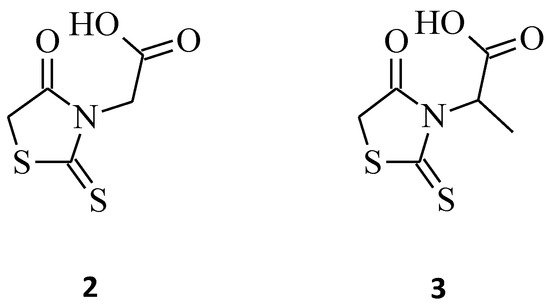
Figure 2. The structures of N-substituted rhodanines.
Furthermore, a structure–activity relationship one was carried out and indicated that, irrespective of the hydrophilic or hydrophobic nature of the groups, the activity decreased with the increase in size [8]. This trend is probably caused by the steric effect that increases with the increasing size of the N-substituents.
Moreover, 3-α-carboxyethyl rhodanine 3 was tested for its anticancer activity against the HeLa (human cervical cancer) cell line, and it turned out to be potent with an IC50 value of 200 µg/mL (Figure 2) [9].
The antiproliferative activity of the N-3-substituted rhodanines was also confirmed by Déliko Dago et al. [10], who evaluated the biological activity of some 3-[4-(arylalkoxy)phenylethyl]-2-thioxo-1,3-thiazolidin-4-one (compound 4) and 3-[2-(4-hydroxyphenyl)ethyl]-2-thioxo-1,3-thiazolidin-4-one (compound 5) against representative tumor cell lines (Figure 3). The results of the survival assays showed that 2-thioxo-1,3-thiazolidin-4-one derivative 4 exhibited selective antitumor activity in the colorectal adenocarcinoma HCT 116 cell line, with an IC50 value of 10 μM, and did not inhibit the growth of normal fibroblasts (IC50 > 25 μM). While compound 5, interestingly, probably due to the presence of the hydroxyl group and lack of bulky substituents, caused a good increase in the antitumor activities, but without selectivity (MDA-MB231 (breast carcinoma) and HCT 116, IC50 2 μM; Caco 2 (colon adenocarcinoma cells), IC50 3 μM).
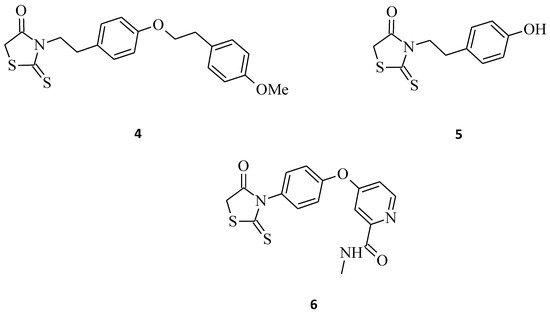
Figure 3. The structures of 3-arylethyl/arylrhodanines.
2.2. 5-Substituted Rhodanine Derivatives
Compound 9, which is 5-{4-[3-(4-methoxy-phenyl)-3-oxo-propenyl]-benzylidene}-2-thioxothiazolidin-4-one (Figure 4), exhibited promising inhibitory activity against the HeLa, HT29 (colorectal adenocarcinomma), A549, and MCF-7 cell lines with the inhibitory concentration (IC50) values of 28.3, 24.5, 26.6, and 28.6 μM, respectively [11]. Moreover, 5-((2-chloro-6,7-dimethoxyquinolin-3-yl)methylene) rhodanine derivative 10 (Figure 4) turned out to be potent against the gastric (HGC), prostate (DU-145), and breast cancer (MCF-7) lines [12]. With some further modifications on pharmacophore, compound 10 could serve as a potential anticancer agent, especially towards the DU-145 and HGC cancer cell lines. By comparing the excellent cytotoxic activity of structures 9 and 10 to some previously described 3-substituted derivatives, it can be concluded that the molecules possessing a free -NH-group in the rhodanine moiety seem to be more potent over the N-CH2-COOH or N-Ph substituted ones. As suggested in the docking studies, this may be connected with the influence of the hydrogen donor group on the active site of the molecular target, such as, for example, the epidermal growth factor receptor (EGFR) [11].
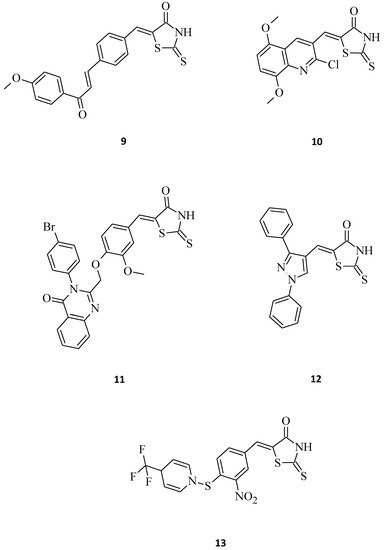
Figure 4. The structures of 5-aryl/heterylmethylidenerhodanines.
El-Sayed et al. [13] synthesized some novel quinazolinone-based rhodanines that were then biologically evaluated for in vitro cytotoxic activity against the human fibrosarcoma cell line HT-1080 and two human leukemia cell lines, namely HL-60 and K562. Amongst them, structure 11, bearing a bulky, hydrophobic substituent at the para position of the quinazolinone 3-phenyl ring, was the most active, showing cytotoxic activity in the low micromolar range (IC50 = 1.2–8.7 µM) towards all the tested cell lines (Figure 4). Its meta-substituted counter partners shown were far less active. Interestingly, normal human skin fibroblasts (AG01523) were not affected by this molecule, which indicates that some rhodanines may be selectively toxic against cancer cells. Another great example of a structure that exhibits selective antitumor activity against selected leukemia and non-small cell lung cancer cell lines is 12. The concentrations of this compound 12 for 50% of the maximal inhibition of the cell proliferation (GI50) were tested, and it turned out to be very potent, especially towards the HOP-92 (non-small cell lung cancer), CCRF-CEM (leukemia), and RPMI-8226 (leukemia) cell lines with GI50 values of 0.62, 2.50, and 2.52 μM, respectively. The described molecule 12 (Figure 4), as a pyrazole-rhodanine derivative with the LC50 > 100 μM indicates the low toxicity of such compounds for normal human cell lines, as required for potential anti-tumor agents [14].
There are also premises in the scientific one regarding some small molecules that might be fairly useful as a starting point to develop novel anticancer agents. As an example, it can be mentioned in structure 13, which was quite toxic against HeLa and Hep cells, with EC50 values of 7.9 and 6.1 μM, respectively (Figure 4) [15].
In comparison, El-Mawgoud [16] synthesized some novel 5-[4-(arylmethylideneamino)-1,5-dimethyl-2-phenyl-1H-pyrazol-3(2H)-ylidene]-2-thioxo-1,3-thiazolidin-4-ones. Compounds 14 and 15 and their cytotoxicity against human breast carcinoma cell line were evaluated (Figure 5). Both of these 5-substituted rhodanines showed high antitumor activity against the cell line MCF-7; however, molecule 14 was more potent than 15 with IC50 values of 7.67 μg/mL and 11.7 μg/mL, respectively. This indicates that increasing the mass of the aryl substituent resulted in a decrease in the cytotoxic activity of the tested compound 15.
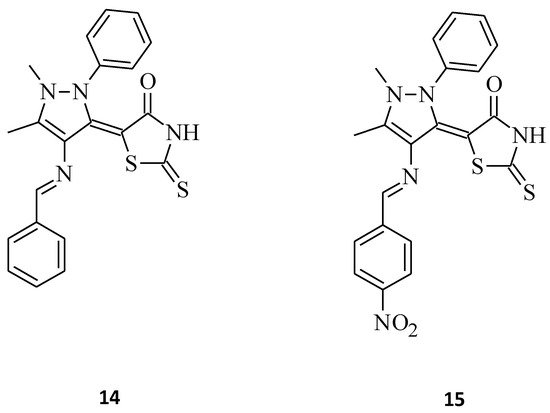
Figure 5. The structures of 5-[4-(phenylmethylideneamino)-1,5-dimethyl-2-phenyl-1H-pyrazol-3(2H)-ylidene]-2-thioxo-1,3-thiazolidin-4-one (14) and 5-[4-({4-nitrophenyl}methylideneamino)-1,5-dimethyl-2-phenyl-1H-pyrazol-3(2H)-ylidene]-2-thioxo-1,3-thiazolidin-4-one (15).
Some new benzimidazole–rhodanine conjugates, 16 and 17, were designed, synthesized, and investigated for their cytotoxic activities against human cancer cell lines, including the human acute leukemia cell line (HL-60), the adenocarcinomic human alveolar basal epithelial cancer cell line (A549), the human lymphoma cancer cell line (Raji), and the human breast cancer cell line (MDA-MB-201) [17]. Compound 16, namely 5-[1-(4-methylbenzyl)-1H-benzo[d]imidazol-2-yl]methylene-2-thioxothiazolidin-4-one, showed excellent inhibitory activity against tested cell lines, with IC50 values of 2.66, 5.31, 4.48, and 6.42 μM, respectively, while the change of the 4-methyl substituent (compound 16) on the phenyl ring to 2-fluoro for compound 17 resulted in a loss of cytotoxic activity towards all cancer cell lines (Figure 6). This may be related to the fact that compounds with electron donating groups showed better Topo II inhibition than those with electron-withdrawing groups [17].
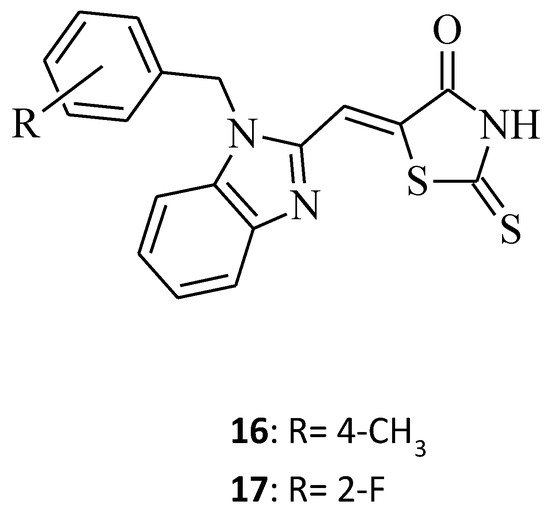
Figure 6. The structure of benzimidazole-rhodanine conjugates.
2.3. 3,5-Disubstituted Rhodanine Derivatives
A new rhodanine analogue bearing 2-piperidine-quinoline scaffold [12], which is compound 18 (Figure 7), was tested on two cancer cell lines, namely the HGC and the MNK 74 (gastric cancer cell line). As with compound 10, the molecule seems to be effective and hopefully, it will be considered as a potential anticancer agent, especially towards gastric cancer, in the future. In turn, structure 19, as a 3,5-disubstituted derivative with a cinnamoyl moiety at the fifth position of the rhodanine nucleus, was screened against MCF-7 breast cancer cells [18] and showed some significant anticancer activity, inhibiting the growth of the cancer cell line by 81% at a concentration 10 µg/mL (Figure 7). According to the analogs of the tested compound 19, shown in [18], the change of the N-3 substitution of the rhodanine ring from 2-chlorophenyl for molecule 19 to 3-cyclohexyl (20) and 3-benzyl (21) (Figure 7) resulted in the inhibitory decline (inhibitory values of 77% and 71%, respectively). This example indicates a trend, showing that increasing the substituent mass in the third position of the rhodanine moiety improves anticancer activity, as it also does amongst the 3,5-disubstituted rhodanine analogues.
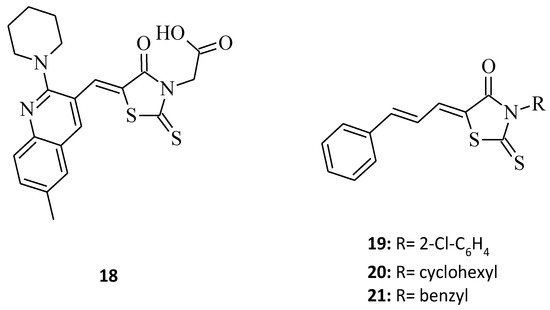
Figure 7. The structures of 3-aryl/alkyl-5-aryl/heterylmethylidenerhodanines.
A good example of the superiority of rhodanines over thiazolidinediones is compound 27, which is a phenyl-substituted triazolothiazolyl-rhodanine derivative [19]. This compound reveals better anticancer properties. This seems to prove that this particular moiety should still be widely researched and used in the development of promising new anticancer agents. The discussed structure showed remarkable cytotoxic activity against two cancer types, namely the hepatocellular carcinoma (HCC) Huh7 and breast cancer MCF-7 cell lines, with IC50 values of 4.67 and 2.30 μM, respectively (Figure 8). At the same time, its analogue, 28, in which the rhodanine moiety was replaced with thiazolidine-2,4-dione, turned out to be non-responsive to the tested cells. It is noteworthy that, according to the results of this study, the lipophilic groups, such as -CH2COOC2H5, introduced on the N-3 position of the rhodanine nucleus, may improve the anticancer activity of the compounds and may increase the permeability of the compound to cells. Lipophilic groups may also have a positive impact when implemented into novel rhodanine derivatives as potential antitumor agents, for the same reasons.
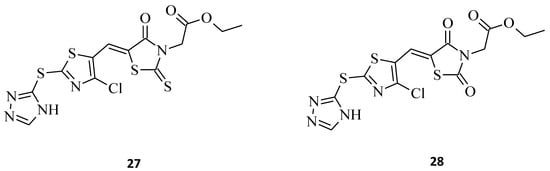
Figure 8. The structures of thiazolyl-rhodanine derivative (27) and its thiazolidine-2,4-dione analogue (28).
The next promising rhodanine compound, with a furochromone scaffold in its structure, is structure 29, which was synthesized and tested for its anticancer properties (Figure 9) [20]. This khellin derivative turned out to be potent on breast cancer cells that originated from different types of tissues, displaying very low EC50 values, especially against the MCF-7 and MDA-MB-231 cell lines (EC50 = 1.732 and 2.912 μM, respectively). In addition, a superior inhibitory effect of growth on Huh7 cells was observed. Based on this form of furochromone, khellin with a lipophilic rhodanine structure, the discovery of even more active molecules slowing down the progression of the tumor cells could be carried out, mainly for novel anti-breast cancer agents.
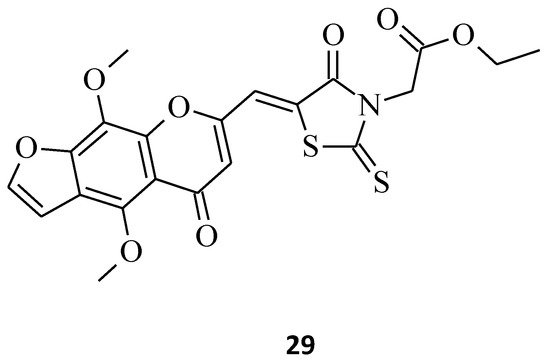
Figure 9. The structure of furochromone derivative.
New 5-(3,5-diaryl-4,5-dihydropyrazol-1-ylmethylene)-2-thioxothiazolidin-4-ones with a diclofenac moiety, namely compound 30 and 31 (Figure 10), have been synthesized and evaluated for their antitumor activities [21]. 2-[2-(2,6-Dichlorophenylamino)-phenyl]-N-{5-[5-(4-methoxyphenyl)-3-naphthalen-2-yl-4,5-dihydropyrazol-1-ylmethylene]-4-oxo-2-thioxothiazolidin-3-yl}-acetamide, 30, was found to be the most active structure possessing substantial activity against all tested human tumor cell lines, with average cell growth indices (GPmean) of 22.40%, whereas molecule 31, being an analogue of 30, with just a 3-phenyl substitution of the pyrazole moiety instead of 3-naphthalene, was a diametrically weaker agent, with average cell growth indices (GPmean) of 99.30%. These rhodanine-pyrazoline hybrid molecules, with a diclofenac moiety after some further modifications on pharmacophore, could potentially serve as a base for designing novel anticancer drugs.
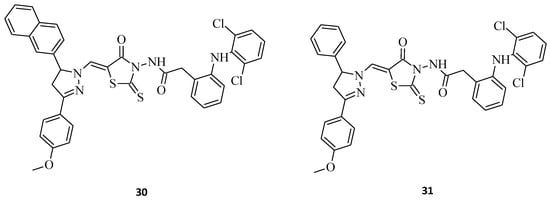
Figure 10. The structures of new rhodanine-pyrazoline hybrid molecules with a diclofenac fragment.
Benzimidazole–rhodanine conjugates 32 and 33 (Figure 11) were synthesized as analogues to the compounds 16 and 17 (Figure 7), being additionally N-3-substituted with acetic moiety [14]. The most potent structure of the discussed compounds was 32, exhibiting excellent cytotoxic activity against the HL-60, MDA-MB-201, Raji, and A549 cancer cell lines, with IC50 values of 0.21, 0.33, 1.23, and 2.67 μM, respectively. The compound was added to the wells at increasing concentrations (0–50 μM). After 48 h, each well was treated with a 20 μL MTT (2.5 mg/mL) solution, and the cells were further incubated at 37 °C for 4 h. In comparison to 17, it seems that acetic moiety is crucial for the cytotoxic effect, at least for the tested cancer cell lines. It is noteworthy that both of the 3,5-disubstituted rhodanines, 32 and 33, displayed significantly better activity than their 5-substituted counterparts from the study. The results show that the introducing of acidic moiety, especially acetic one, at the third position of the rhodanine ring may have a significant impact on the potential anticancer activity of the desired compounds.
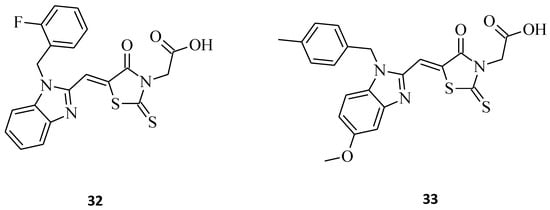
Figure 11. The structures of benzimidazole–rhodanine conjugates.
Another indisputable piece of evidence confirming the superiority of 3,5-disubstituted structures over their 3-substituted rhodanine counterparts, with regard to their anticancer properties, is compound 34. This 3-α-carboxy ethyl-5-benzylidene rhodanine derivative caused inhibition of HeLa cancer cell growth by 52% (Figure 12), while 3 (Figure 2) was less effective against the tested HeLa cells, with an inhibitory percentage of 14.28% [9]. When comparing these two structures, it is clear that the introduction of 4-methoxy benzylidene moiety for 34 increased its cytotoxicity levels significantly towards the tested HeLa cancer cells.
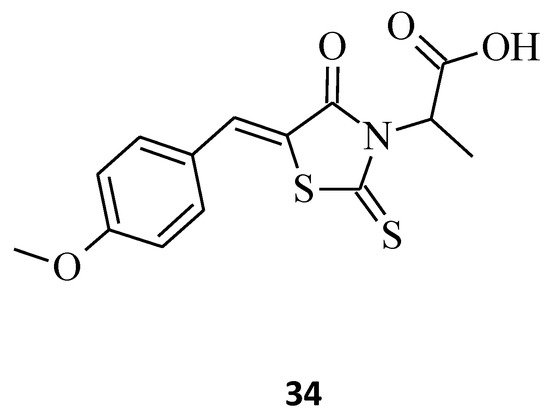
Figure 12. The structure of 5-{[(4-methoxyphenyl)methylidene]-4-oxo-2-thioxo-1,3-thiazolidin-3-yl}propanoic acid.
Novel 3-(4-Arylmethylamino)butyl-5-arylidene-rhodanine, 35, was synthesized [22], and its antitumor activity was tested. This structure exhibited promising antitumor effects in the HuH7 D12, HaCat, and MDA-MBD 231 cell lines, with IC50 values below 10 μM (Figure 13). It is worth emphasizing that compound 35, while being potent against cancer cell lines, did not inhibit the growth of normal fibroblasts (IC50 > 25 μM).
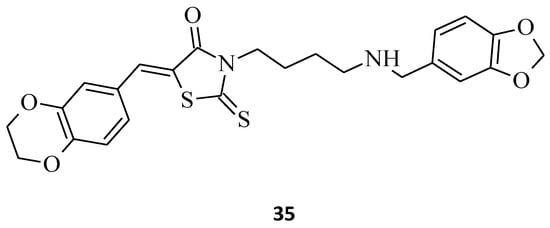
Figure 13. The structure of 3-(4-arylmethylamino)butyl-5-arylidenerhodanine.
Kryshchyshyn et al. introduced some new pyrrolidinedione-thiazolidinone hybrids, 36 and 37 (Figure 14), and then tested these 5-ylidene-3-(1-aryl-pyrrolidine-2,5-dione)-rhodanines towards selected cell lines for their antileukemic properties [23]. Both compounds inhibited Dami cell line growth by more than 50%, and 36 was the more potent of the two (Dami cell line growth = 35.10%). In turn, structure 37 turned out to be more active against HL-60 cells, with an inhibitory value of almost 60%. Based on the presented data, one could say that compounds 36 and 37 possess satisfactory toxicity levels on leukemia cell lines and might be used for the drug-like molecules.
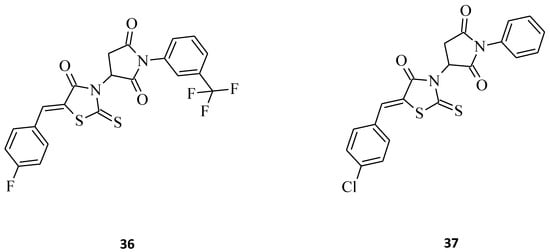
Figure 14. The structures of pyrrolidinedione-thiazolidinone hybrids.
Selected rhodanine-3-carboxylic acid derivative, 38, was synthesized and its cytotoxicity against human ovarian carcinoma A2780 and A2780cisR-cells has been determined [24]. Structure 38, namely 4-[5-(4′-N,N-dimethylaminobenzylidene)-rhodanine]-butyric acid, displayed excellent anticancer activity, with IC50 = 4.4 and 3.3 μM towards both tested cell lines, A2780 and A2780cisR, respectively (Figure 15). Interestingly, the selected compound 38 was much more cytotoxic than cisplatin in both cancer cell lines. Phenothiazine, chalcone, and rhodanine moieties that are pharmacologically active were presented in the hybrid molecule 39 and seem to act synergetically when evaluated for their antiproliferative activity against K562 cancer cell lines (Figure 15) [25].
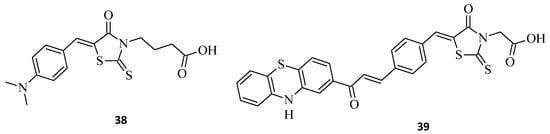
Figure 15. The structures of 5-arylidenerhodanine-3-carboxylic acids.
Buzun et al. [26] designed and synthesized a series of new 5-[(Z,2Z)-2-chloro-3-(4-nitrophenyl)-2-propenylidene]-thiazolidinones, which are a combination of a thiazolidinone core and a structural fragment of the ciminalum, namely(2Z)-2-chloro-3-(4-nitrophenyl)prop-2-enal. Ciminalum is an active Gram-positive and Gram-negative antimicrobial factor [27]. Amongst these hybrid compounds, 3-{5-[(Z,2Z)-2-chloro-3-(4-nitrophenyl)-2-propenylidene]-4-oxo-2-thioxothiazolidin-3-yl}propanoic acid, 40 (Figure 16), displayed the best antimitotic activity, with mean GI50 values of 1.57 μM and a certain sensitivity range towards the leukemia (MOLT-4, SR), colon cancer (SW-620), CNS cancer (SF-539), melanoma (SK-MEL-5), gastric cancer (AGS), human colon cancer (DLD-1), and breast cancers (MCF-7, MDA-MB-231) cell lines. Structure 41, being a p-hydroxyphenyl derivative was also very effective, while the absence of a substituent in the C-3 position of the rhodanine moiety (42), or an additional ciminalum fragment (43), led to decrease in anticancer cytotoxicity (Figure 16). Both compound 40 and compound 41 had low toxicity levels towards normal human blood lymphocytes and a broad range of therapeutic effects. These data suggest that the presence of a ciminalum moiety in the C-5 position of the 2-thioxo-4-thiazolidinone ring is a very interesting possibility for designing novel and potentially active agents, as high cytotoxicity of the tested 5-[(Z,2Z)-2-chloro-3-(4-nitrophenyl)-2-propenylidene]-2-thioxo-4-thiazolidinone-3-carboxylic acids against several cancer cell lines have been established.
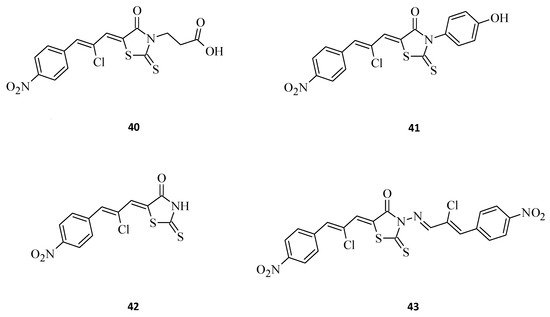
Figure 16. The structures of new ciminalum–thiazolidinone hybrid molecules.
Zhou et al. [28] combined the cores of a 2-thioxo-4-thiazolidinone moiety, a, b-unsaturated ketones, and acrylamide derivatives to design new microtubule-interacting agents as potentially active antiproliferative compounds against different cancer cells. (Z)-2-(5-(4-(dimethylamino) benzylidene)-4-oxo-2-thioxothiazolidin-3-yl)-N-phenylacetamide, 44 (Figure 17), displayed the best antiproliferative activity towards A549 (IC50 = 7 μM) cancer cells, comparable to that achieved with gefitinib (IC50 = 5.89 μM). Moreover, molecule 44 turned out to be only weakly cytotoxic against NRK-52E cells, with IC50 = 14.7 μM, while promoting microtubule protofilament assembly, leading to a reduction in microtubule density and disordered networks. It seems that a bulky steric-hindering moiety at the para position favors the good bioactivity of modified (Z)-2–(5-benzylidene-4-oxo-2-thioxothiazolidin-3-yl)-N-phenylacetamide derivatives, according to compound 44. These results might help with developing novel microtubule-stabilizing structures, which are potent in the treatment of cancer.
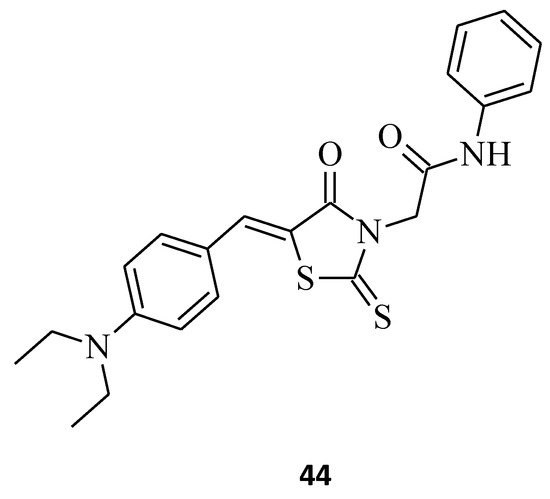
Figure 17. The structure of (Z)-2-(5-benzylidene-4-oxo-2-thioxothiazolidin-3-yl)-N-phenylacetamide.
Last, but not least, rhodanine-oleanolic acid derivatives, 45 and 46 [29], had a significant inhibitory effect on some breast cancer (45) and ovarian cancer (46) cell lines (Figure 18). However, any tendency between the cytotoxic effects for different substituents of these oleanolic derivatives, including 7 and 8 (Figure 4) compounds and cancer cell lines, is difficult to determine.
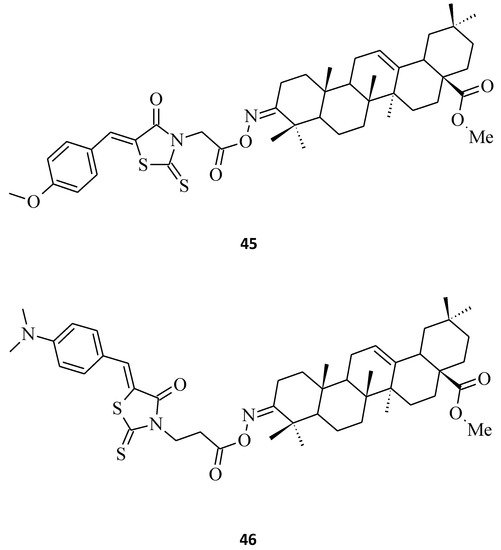
Figure 18. The structures of 3-O-acyloleanolic acid derivatives with rhodanine core.
Summarizing the structure–activity relationship analysis, the following trend can be observed. The introduction of small substituents in position 3 of the (2-thioxothiazolidin-3-yl)acetic acid derivatives (compounds 2 and 3) improves the activity against the leukemia cell line K562. However, the enlarging of the substituents in this position (ex. isopropyl, carboxyethyl, or benzyl) was unfavorable for antiproliferative activity against K562 (Figure 19).
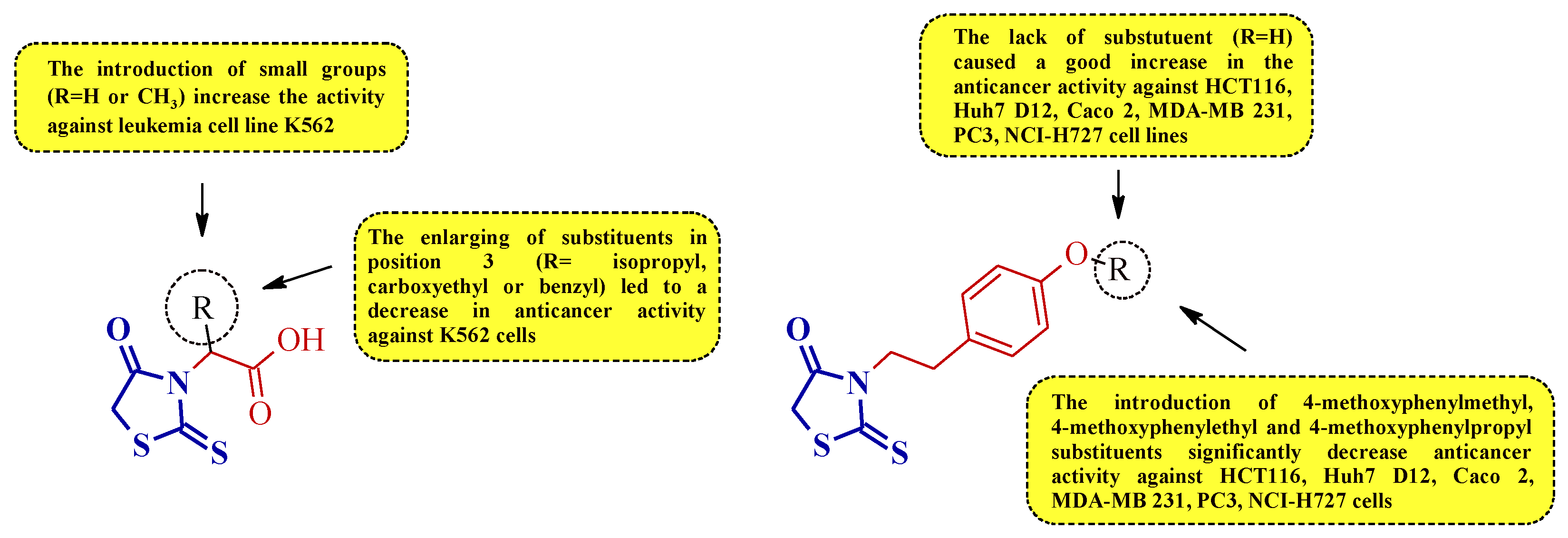
Figure 19. The structure–activity relationship for some 3-substituted rhodanines with anticancer properties against leukemia, colorectal, prostate, breast, hepatocellular, and lung carcinoma cells.
A similar trend was observed for compounds 5 and 4. Expanding the substituent by the 4-methoxyphenylalkyl groups of compound 5 decreased the anticancer activity against some leukemia, colorectal, prostate, breast, hepatocellular, and lung cancer cell lines (Figure 19).
It is notable that the presence of heteryl moiety was more preferable for good anticancer activity than aryl substituent in 5-substituted rhodanines (Figure 20).
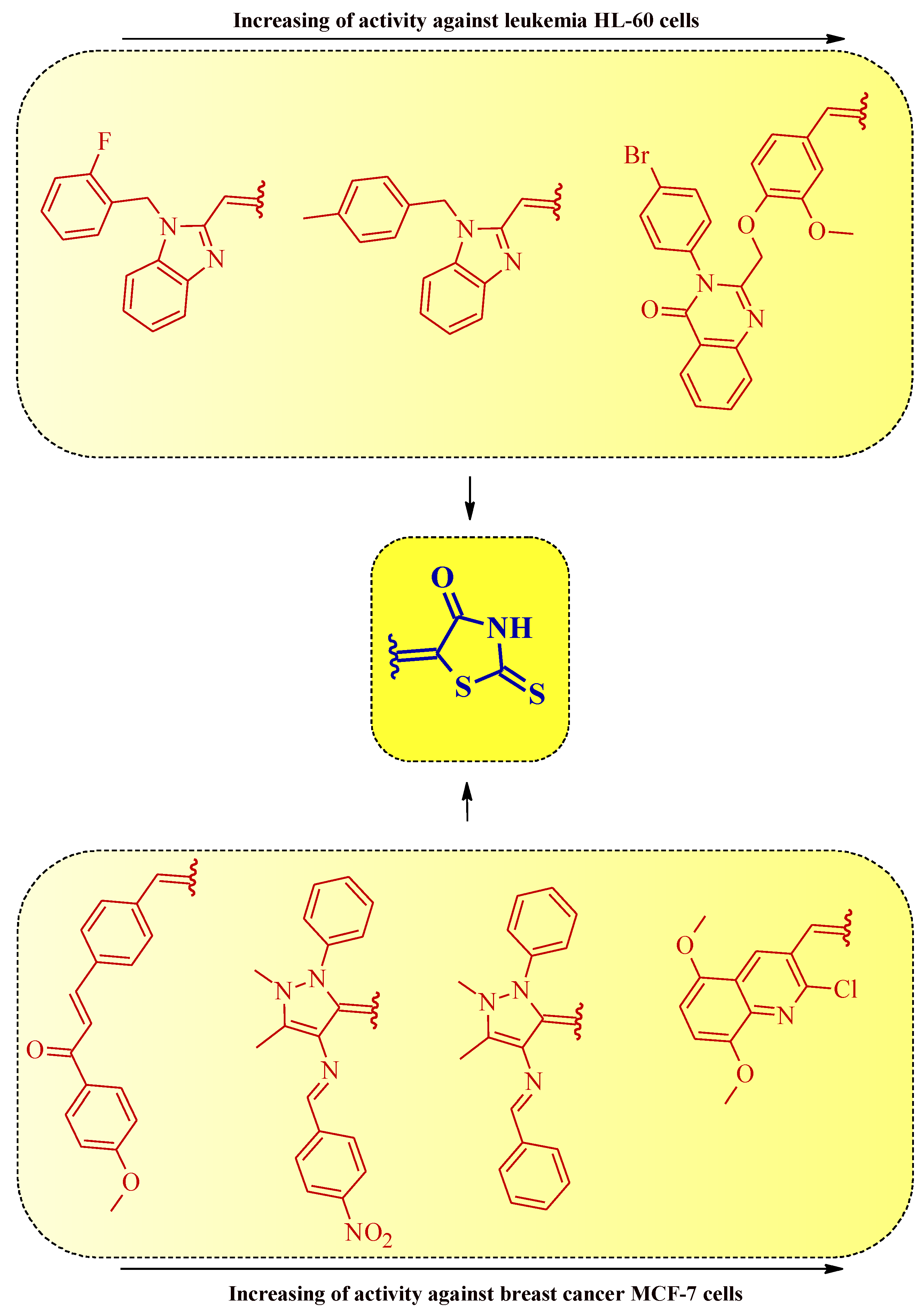
Figure 20. The structure–activity relationship for some 5-substituted rhodanines with anticancer properties against leukemia and breast cancer cells.
It is worth noticing that the introduction of simultaneous substituents at positions 3 and 5 of the rhodanine system generally increases the anticancer activity in comparison with the 3- or 5-monosubstituted rhodanine derivatives (Figure 21).
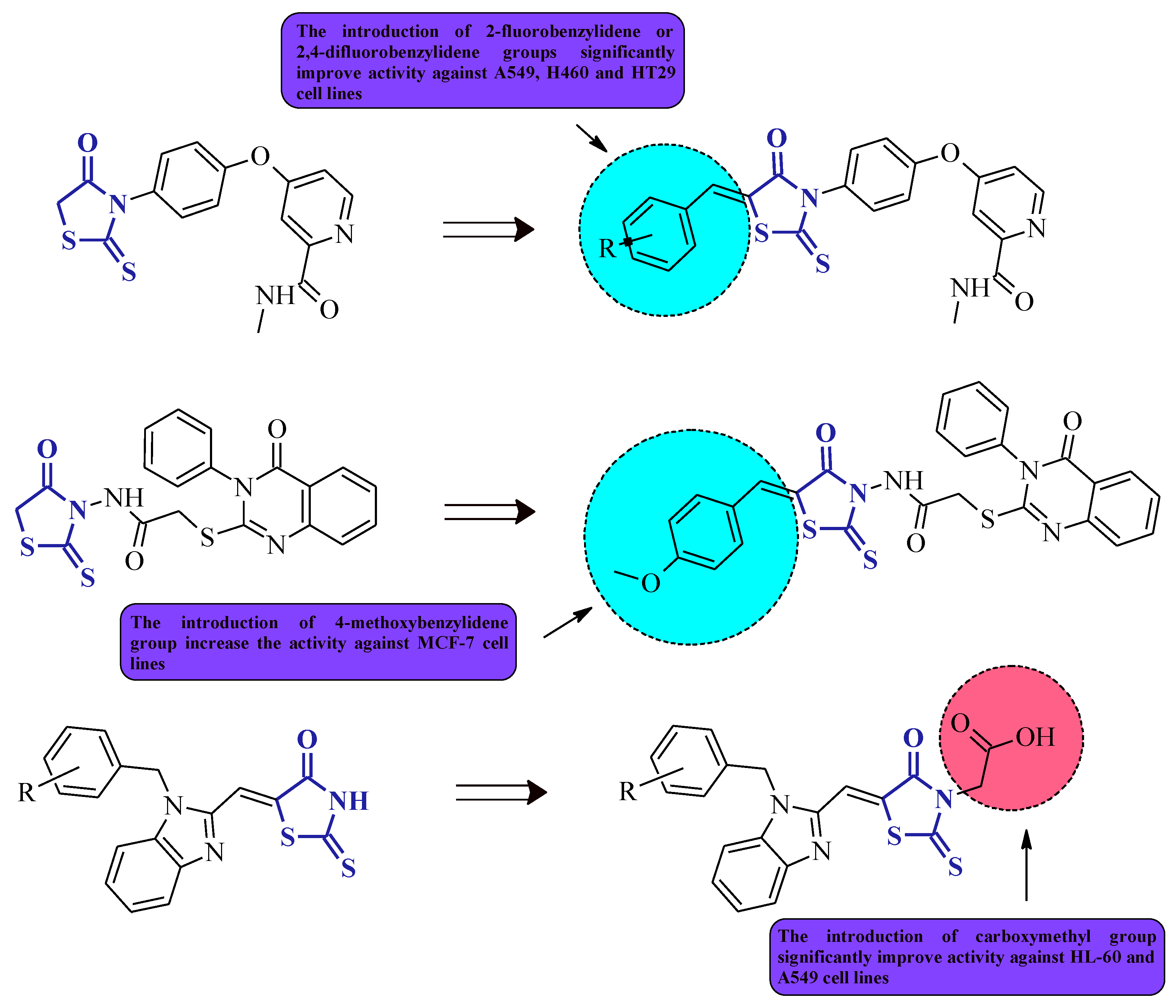
Figure 21. The structure–activity relationship for some 3,5-disubstituted rhodanines with anticancer properties.
References
- Li, H.; Yang, J.; Ma, S.; Qiao, C. Structure-based design of rhodaninebased acylsulfonamide derivatives as antagonists of the anti-apoptotic Bcl-2 protein. Bioorg. Med. Chem. 2012, 20, 4194–4200.
- Bernardo, P.; Sivaraman, T.; Wan, K.; Xu, J.; Krishnamoorthy, J.; Song, C.; Tian, L.; Chin, J.; Lim, D.; Mok, H.; et al. Structural insights into the design of small molecule inhibitors that selectively antagonize Mcl-1. J. Med. Chem. 2010, 53, 2314–2318.
- Chandrappa, S.; Kavitha, C.; Shahabuddin, M.; Vinaya, K.; Kumar, C.; Ranganatha, S.; Raghavan, S.; Rangappa, K. Synthesis of 2-(5-((5-(4-chlorophenyl)furan-2-yl)methylene)-4-oxo-2-thioxothiaz-olidin-3-yl)acetic acid derivatives and evaluation of their cytotoxicity and induction of apoptosis in human leukemia cells. Bioorg. Med. Chem. 2009, 17, 2576–2584.
- Ravi, S.; Chiruvella, K.; Rajesh, K.; Prabhu, V.; Raghavan, S. 5-Isopro-pylidene-3-ethyl rhodanine induce growth inhibition followed by apoptosis in leukemia cells. Eur. J. Med. Chem. 2010, 45, 2748–2752.
- Ahn, J.; Kim, S.; Park, W.; Cho, S.; Ha, J.; Kim, S.; Kang, S.; Jeong, D.; Jung, S.; Lee, S.; et al. Synthesis and biological evaluation of rhodanine derivatives as PRL-3 inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 2996–2999.
- Moorthy, B.T.; Ravi, S.; Srivastava, M.; Chiruvella, K.K.; Hemlal, H.; Joy, O.; Raghavan, S.C. Novel rhodamine derivatives induce growth inhibition followed by apoptosis. Bioorg. Med. Chem. Lett. 2010, 20, 6297–6301.
- Nguyen, C.T.; Nguyen, Q.T.; Dao, P.H.; Nguyen, T.L.; Nguyen, P.T.; Nguyen, H.H. Synthesis and cytotoxic activity against K562 and MCF7 cell lines of some N-(5-arylidene-4-oxo-2-thioxothiazolidin-3-yl)-2-((4-oxo-3-phenyl-3,4-dihydroquinazoline-2-yl)thio)acetamide compounds. J. Chem. 2019, 2019, 1492316.
- Ali Muhammad, S.; Ravi, S.; Thangamani, A. Synthesis and evaluation of some novel N-substituted rhodanines for their anticancer activity. Med. Chem. Res. 2016, 25, 994–1004.
- Kaveri, S.; Ravi, S. Synthesis, antibacterial activity against MRSA, and in vitro cytotoxic activity against HeLa cell lines of novel 3-α-carboxy ethyl-5-benzylidene rhodanine derivatives. Res. Chem. Intermed. 2015, 41, 1011–1021.
- Dago, C.D.; N’Ta Ambeu, C.; Coulibaly, W.K.; Békro, Y.-A.; Mamyrbekova-Bekro, J.A.; Le Guevel, R.; Corlu, A.; Bazureau, J.-P. Investigation on the synthesis of new 3--2-thioxo-1,3-thiazolidin-4-ones and their biological evaluation against cancer cells. Chem. Heterocycl. Compd. 2017, 53, 341–349.
- Alegaon, S.G.; Alagawadi, K.R.; Vinod, D.; Unger, B.; Khatib, N.A. Synthesis, pharmacophore modeling, and cytotoxic activity of 2-thioxothiazolidin-4-one derivatives. Med. Chem. Res. 2014, 23, 5160–5173.
- Ramesh, V.; Rao, B.A.; Sharma, P.; Swarna, B.; Thummuri, D.; Srinivas, K.; Naidu, V.G.M.; Rao, V.J. Synthesis and biological evaluation of new rhodanine analogues bearing 2-chloroquinoline and benzoquinoline scaffolds as anticancer agents. Eur. J. Med. Chem. 2014, 83, 569–580.
- El-Sayed, S.; Metwally, K.; El-Shanawani, A.A.; Abdel-Aziz, L.; Pratsinis, H.; Kletsas, D. Synthesis and anticancer activity of novel quinazolinone-based rhodanines. Chem. Cent. J. 2017, 11, 102.
- Insuasty, A.; Ramírez, J.; Raimondi, M.; Echeverry, C.; Quiroga, J.; Abonia, R.; Nogueras, M.; Cobo, J.; Rodríguez, M.V.; Zacchino, S.A.; et al. Synthesis, antifungal and antitumor activity of novel (Z)-5-hetarylmethylidene-1,3-thiazol-4-ones and (Z)-5-ethylidene-1,3-thiazol-4-ones. Molecules 2013, 18, 5482–5497.
- Strittmatter, T.; Bareth, B.; Immel, T.A.; Huhn, T.; Mayer, T.U.; Marx, A. Small molecule inhibitors of human DNA polymerase λ. ACS Chem. Biol. 2011, 6, 314–319.
- El-Mawgoud, H.K.A. Synthesis, in vitro cytotoxicity and antimicrobial evaluations of some novel thiazole based heterocycles. Chem. Pharm. Bull. 2019, 67, 1314–1323.
- Li, P.; Zhang, W.; Jiang, H.; Li, Y.; Dong, C.; Chen, H.; Zhang, K.; Du, Z. Design, synthesis and biological evaluation of benzimidazole-rhodanine conjugates as potent topoisomerase II inhibitors. Med.Chem.Comm. 2018, 9, 1194–1205.
- Mandal, S.P.; Mithuna; Garg, A.; Sahetya, S.S.; Nagendra, S.R.; Sripad, S.H.; Manjunath, M.M.; Sitaram; Soni, M.; Nasir Baig, R.; et al. Novel rhodanines with anticancer activity: Design, synthesis and CoMSIA study. RSC Adv. 2016, 6, 58641–58653.
- Özen, C.; Ceylan-Ünlüsoy, M.; Aliary, N.; Öztürk, M.; Bozdağ-Dündar, O. Thiazolidinedione or rhodanine: A study on synthesis and anticancer activity comparison of novel thiazole derivatives. J. Pharm. Pharm. Sci. 2018, 20, 415–427.
- Demir, S.; Özen, C.; Ceylan-Ünlüsoy, M.; Öztürk, M.; Bozdağ-Dündar, O. Novel furochromone derivatives: Synthesis and anticancer activity studies. J. Heterocycl. Chem. 2019, 56, 1341–1351.
- Shepeta, Y.; Lozynskyi, A.; Tomkiv, Z.; Grellier, P.; Lesyk, R. Synthesis and evaluation of the biological activity of rhodanine-pyrazoline hybrid molecules with 2-(2,6-dichlorophenylamino)-phenylacetamide fragment. Biopolym. Cell 2020, 36, 133–145.
- Dago, C.D.; Ambeu, C.N.; Coulibaly, W.-K.; Békro, Y.-A.; Mamyrbékova, J.; Defontaine, A.; Baratte, B.; Bach, S.; Ruchaud, S.; Guével, R.L.; et al. Synthetic development of new 3-(4-arylmethylamino)butyl-5-arylidene-rhodanines under microwave irradiation and their effects on tumor cell lines and against protein kinases. Molecules 2015, 20, 12412–12435.
- Kryshchyshyn, A.; Kaminskyy, D.; Roman, O.; Kralovics, R.; Karpenko, A.; Lesyk, R. Synthesis and anti-leukemic activity of pyrrolidinedione-thiazolidinone hybrids. Ukr. Biochem. J. 2020, 92, 108–119.
- Stawoska, I.; Tejchman, W.; Mazuryk, O.; Lyčka, A.; Nowak-Sliwinska, P.; Żesławska, E.; Nitek, W.; Kania, A. Spectral characteristic and preliminary anticancer activity in vitro of selected rhodanine-3-carboxylic acids derivatives. J. Heterocycl. Chem. 2017, 54, 2889–2897.
- Muhammad, S.A.; Ravi, S.; Thangamani, A.; Chandrasekaran, B.; Ramesh, M. Synthesis, antiproliferative activity and docking study of novel rhodanine derivatives as Bcr-Abl T1351 inhibitors. Res. Chem. Intermed. 2017, 43, 5871–5887.
- Buzun, K.; Kryshchyshyn-Dylevych, A.; Senkiv, J.; Roman, O.; Gzella, A.; Bielawski, K.; Bielawska, A.; Lesyk, R. Synthesis and anticancer activity evaluation of 5--4-thiazolidinones. Molecules 2021, 26, 3057.
- Antypenko, L.; Gladysheva, S. Development, and validation of UV-spectrophotometric determination of ciminalum in drug. Recipe 2017, 20, 153–160.
- Zhou, X.; Liu, J.; Meng, J.; Fu, Y.; Wu, Z.; Ouyang, G.; Wang, Z. Discovery of facile amides-functionalized rhodanine-3-acetic acid derivatives as potential anticancer agents by disrupting microtubule dynamics. J. Enzyme Inhib. Med. Chem. 2021, 36, 1996–2009.
- Kaminskyy, D.; Bednarczyk-Cwynar, B.; Vasylenko, O.; Kazakova, O.; Zimenkovsky, B.; Zaprutko, L.; Lesyk, R. Synthesis of new potential anticancer agents based on 4-thiazolidinone and oleanane scaffolds. Med. Chem. Res. 2012, 21, 3568–3580.
More
Information
Subjects:
Chemistry, Medicinal
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.5K
Revisions:
2 times
(View History)
Update Date:
20 Jun 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No