Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Wu, J.; , .; Chen, H.; Lu, G. Insights into N-Glycanase 1. Encyclopedia. Available online: https://encyclopedia.pub/entry/24131 (accessed on 27 July 2026).
Wu J, , Chen H, Lu G. Insights into N-Glycanase 1. Encyclopedia. Available at: https://encyclopedia.pub/entry/24131. Accessed July 27, 2026.
Wu, Jin, , Hongping Chen, Guanting Lu. "Insights into N-Glycanase 1" Encyclopedia, https://encyclopedia.pub/entry/24131 (accessed July 27, 2026).
Wu, J., , ., Chen, H., & Lu, G. (2022, June 16). Insights into N-Glycanase 1. In Encyclopedia. https://encyclopedia.pub/entry/24131
Wu, Jin, et al. "Insights into N-Glycanase 1." Encyclopedia. Web. 16 June, 2022.
Copy Citation
The cytosolic PNGase (peptide:N-glycanase), also known as peptide-N4-(N-acetyl-β-glucosaminyl)-asparagine amidase, is a well-conserved deglycosylation enzyme (EC 3.5.1.52) which catalyzes the non-lysosomal hydrolysis of an N(4)-(acetyl-β-d-glucosaminyl) asparagine residue (Asn, N) into a N-acetyl-β-d-glucosaminyl-amine and a peptide containing an aspartate residue (Asp, D). This enzyme (NGLY1) plays an essential role in the clearance of misfolded or unassembled glycoproteins through a process named ER-associated degradation (ERAD). Accumulating evidence also points out that NGLY1 deficiency can cause an autosomal recessive (AR) human genetic disorder associated with abnormal development and congenital disorder of deglycosylation.
N-glycanase
NGLY1
PNGase
1. The Discovery of Peptide:N-Glycanase
The glyco-peptidase, partially purified from the seed emulsion of a fruit plant (almond, Prunus dulcis), was first reported in 1977 in order to show its enzymatic activity to cleave β-aspartyl-glycosyl-amine linkages in glyco-peptides [1]. Later, this glyco-peptidase was discovered in several other plants, including jack bean (Canavalia ensiformis) [2], split pea (Pisum sativum) [3], thale cress (Arabidopsis thaliana) [4], lentil (Lens culinaris), pinto bean (Phaseolus vulgaris), lima bean (Phaseolus limensis), barley (Hordeum vulgare) and wheat (Triticum vulgare) [3]. This implied that the existence of this specific glyco-peptidase might be ubiquitous in the plant kingdom. In 1978, some basic enzymatic characters of this new glyco-peptidase such as Km value, optimum pH, and inhibitors were revealed [5]. Since the enzyme could only hydrolyze N-glyco-peptides longer than three amino acids [5], it was named peptide:N-glycosidase (PNGase). It was reported in 1981 that PNGase (PNGase A for almond) could highly efficiently cleave short ovalbumin- and bromelain-derived N-glyco-peptides which were modified with a high content of mannose and complex oligosaccharides [6]. Interestingly, it was discovered in the same year that the carbohydrate removal of pepsin-digested glycoproteins by PNGase was almost three times higher than that of the intact untreated proteins [7]. This implied that the complex oligosaccharides might hinder the accessibility of PNGase to the cleavage site of the target glycoproteins in their native state. Treating glycoproteins with heat in sodium dodecyl sulfate (SDS) or high concentrations of chao-tropic salts such as NaSCN and NaClO4 with disulfide bond reducing agent β-mercapto-ethanol (BME) to denature the native structures, more N-glycans were released from the denatured and unfolded proteins upon treatment with PNGase [8]. In 1984, the enzymatic activity of PNGase was discovered in the Gram-negative bacterial pathogen for meningitis and septicemia, Flavobacterium meningosepticum, and was named PNGase F [9]. This was the discovery of PNGase in prokaryotes. More importantly, in 1989, free di-N-acetyl-chitobiose based oligosaccharides were detected in the extract of eggs of trout (Plecoglossus altivelis) [10], dace (Tribolodon hakonensis) and flounder (Paralichthys olivaceus) [11]. Since di-N-acetyl-chitobiose was mainly produced from N-linked glycoproteins by PNGase, it is quite reasonable to infer the presence of PNGase in the animal kingdom. This led to the discovery of the Peptide:N-Glycosidase in the embryos of Madaka fish (Oryzias latipes) in 1991 [12]. Two years later, PNGase was discovered in mouse (L-929, BALB-3T3 and P3X63-Ag8.U1) and human (TIG-3S) cell lines [13]. In 1997 and 1998, PNGase was identified in two fungi, Aspergillus tubigensis [14] and Saccharomyces cerevisiae [15], respectively. Up to now, the existence of PNGase has been found in the plant, prokaryotes, fungi, and animal kingdoms (Figure 1). ThisPNGase was named Png1 in Arabidopsis thaliana, Png1 in yeast, png-1 Caenorhabditis elegans, Pngl in fruit fly, Ngly1 in mice and rat, and NGLY1 in humans. For readability, the gene is termed NGLY1/Ngly1 in the whole article.
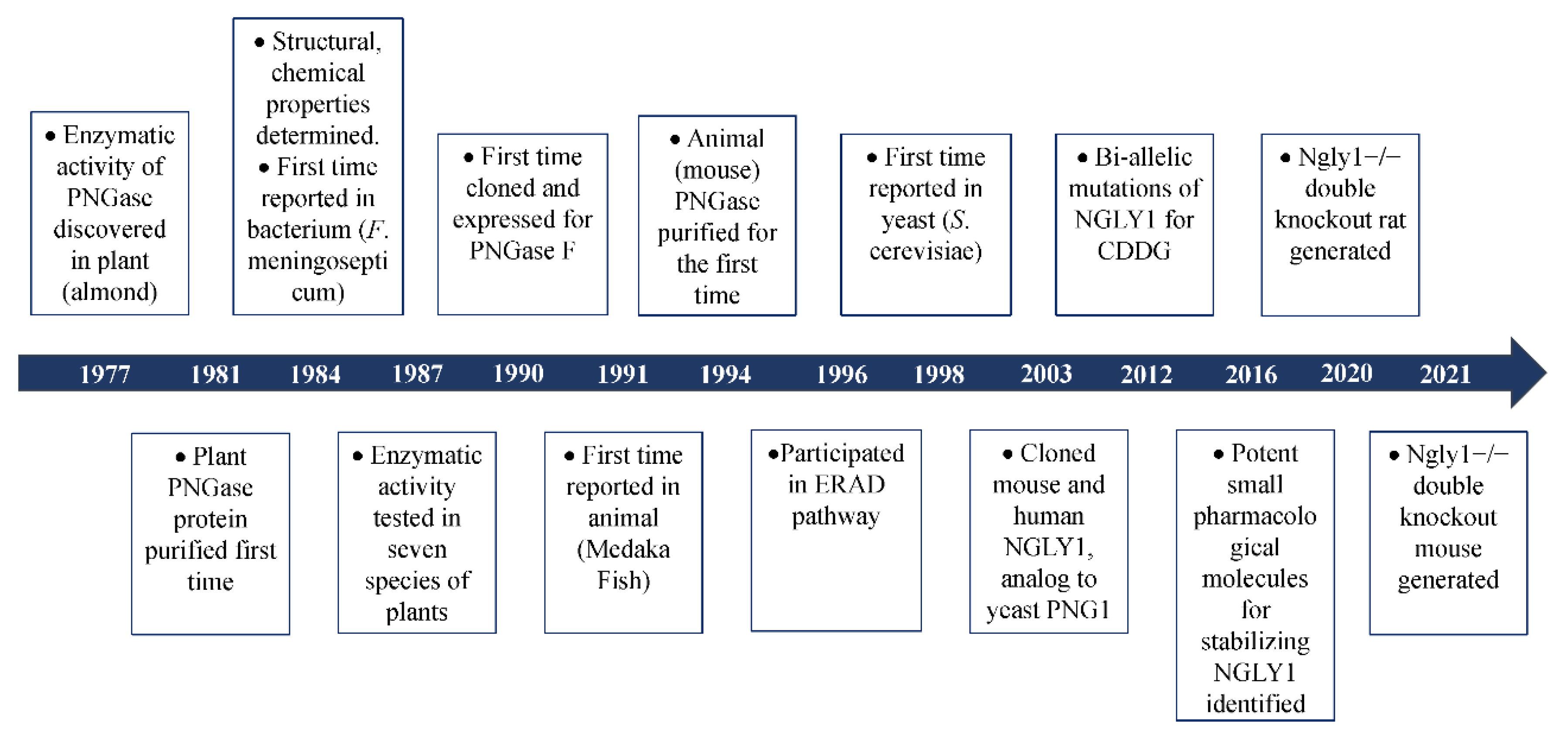
Figure 1. Brief history of the discovery of PNGase.
Based on experimental and bio-informatic analysis, Ngly1 is universally expressed in almost all living cells. Loss-of-functional (LoF) mutations of Ngly1 might be detrimental to the deglycosylation process. In 2012, bi-allelic compound mutations of NGLY1 were identified by whole-exome sequencing (WES) in a boy with congenital anomalies and/or intellectual disabilities [16]. This was the first time that NGLY1 was associated with a specific disorder. Since the disorder was caused by pathologic mutations in the specific enzyme responsible for deglycosylation, the disease was named congenital disorder of deglycosylation (CDDG) or NGLY1-congenital disorder of deglycosylation (NGLY1-CDDG; OMIM#615273). With the discovery of mutations in NGLY1 for CDDG [16][17][18][19][20], more and more researches are concentrated on the molecular mechanisms of NGLY1 in the process of deglycosylation (Figure 1).
2. Different Functional Pathways Participated in by NGLY1
It was widely accepted that NGLY1 played an important role in the ER-associated degradation pathway to remove misfolded glycosylated proteins which were retro-translocated from ER lumen to the cytosol [17][21][22]. Under conditions of NGLY1 deficiency, the misfolded glycoproteins would be accumulated to form insoluble aggregates [17] which could inhibit the activity of 26S and lead to ER stress and cell death [23]. It seemed quite likely that NGLY1 might be involved in a general mechanism to deglycosylate misfolded glycoproteins through the ERAD pathway. However, it has been reported that some proteins could be degraded regardless of glycosylation status [24][25]. In cells with NGLY1 deficiency from Drosophila melanogaster [26] and rat [27], mouse [28] and humans [29], no evidences of ER stress were detected, which was often observed in cells with impaired ERAD function. Several in vivo studies showed that the degradation rate of the majority of ERAD substrates studied was not profoundly altered by a large number of conditions that induced Ngly1 inhibition, such as deletion of the corresponding gene in yeast [30], or use of specific RNAi (RNA interferences) [25], or the inhibitor Z-VAD-FMK (a caspase inhibitor) [31]. This indicated that NGLY1 might play diversified roles in living cells, in addition to being involved in ERAD (Figure 2).
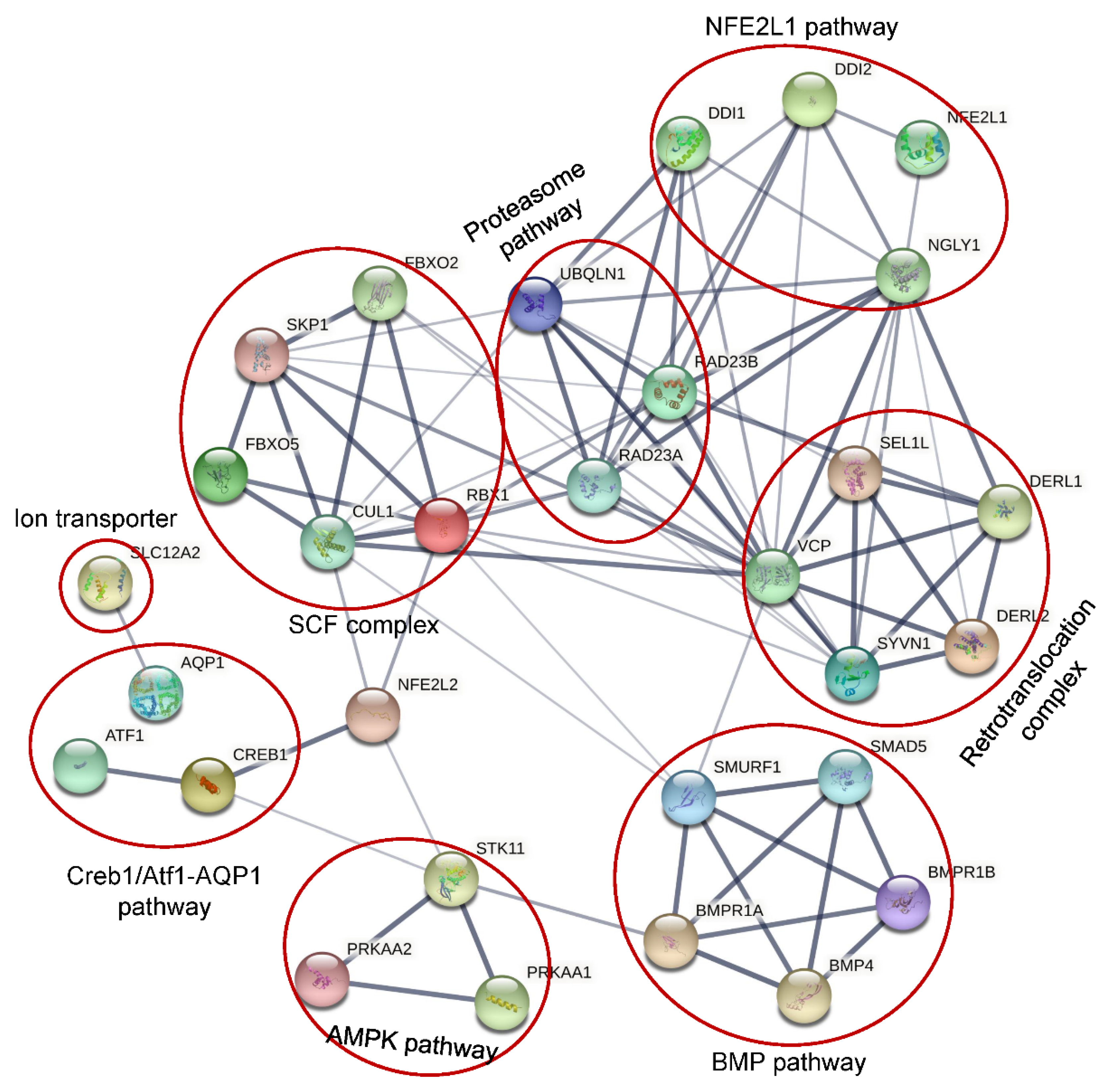
Figure 2. Protein network involving NLGY1 by STRING.
2. 1. NFE2L1 Pathway
NFE2, like bZIP transcription factor 1 (NFE2L1, also called NRF1) is a transcription factor belonging to the CNC-bZIP family [32]. NFE2L1 is involved in regulation of many cellular functions, such as oxidative stress response, differentiation, inflammatory response, and metabolism. NFE2L1 was ubiquitously expressed and could be induced by cellular stresses such as oxidative stress, ER stress, and inflammation. Under a normal physiological state, the N-glycosylated non-active NFE2L1 was retro-translocated into cytosol for ERAD-dependent degradation by the proteasome [33]. Under cellular stimuli for inhibition of the proteasome function, the retro-translocated NFE2L1 escaped the ERAD and the N-glycans were removed by non-ER bound NGLY1 to make the amino acid transition from Asn (N) to Asp (D) [34]. The deglycosylated NFE2L1 was further cleaved at the Leu104 residue by DNA damage inducible 1 homologs (DDI 1 or DDI 2) to release the ER-bound NFE2L1 from the ER membrane [34][35][36]. The processed NFE2L1 (p110) then entered into the nucleus to activate expression of a subset of proteasome subunits to promote the proteasome function in order to alleviate cellular stresses [32]. However, in NGLY1 deficient cells, the N-linked glycans could not be released, which inhibited the cleavage and activation of NFE2L1. It has been identified that NFE2L1 is highly expressed in the brain, heart, kidney, skeletal muscle, and fat [37]. Disruption of the NFE2L1 pathway caused by NGLY1 mutations might be related to the neurological, renal, skeletal and ophthalmological phenotypes of patients with NGLY1 deficiency.
2.2. Creb1/Atf1-AQP Pathway
It has been reported that, regardless of the N-glycanase enzymatic activity, NGLY1 deficiency could decrease transcriptionally the levels of multiple aquaporins (AQPs) by 50–60% in human and mouse cells, indirectly through transcription factors Atf1/Creb1 [28]. Since aquaporins function as a water transmembrane transporter, their decrease could partly explain poor or absent tear production, dry mouth, reduced saliva production [38] and constipation [39].
2.3. BMP Pathway
In Drosophila, loss of Ngly1 could result in developmental midgut defects, which were similar to the deficiency of BMP signaling [40]. Later, Ngly1 was found to colocalize with endoplasmic reticulum via VCP (p97) to promote the retro-translocation and de-glycosylate the misfolded BMP4 for proteasome degradation, which could increase the efficient traffic of properly-folded BMP4 to its target compartment through the secretary pathway [41]. It has been reported that BMP4 is essential for mesoderm development, limb formation, tooth development, bone induction, nephric duct formation, renal system segmentation and aortic valve morphogenesis [42][43][44]. Mutations of BMP4 could result in eye and brain developmental anomalies [42][45][46], which overlap with NGLY1 deficiency.
2.4. AMPK Pathway
In some NGLY1 deficiency patients, altered muscle and liver mitochondrial amount, function and impaired physiology were identified, which could be rescued by restoration of NGLY1 expression, which confirmed the direct relationship of NGLY1 with mitochondrial function [47]. Recently, it has been reported that NGLY1 deficiency could severely reduce the expression and phosphorylation of AMP-activated protein kinase α (AMPKα) in Drosophila larval intestine, mouse embryonic fibroblasts and patient-derived fibroblasts, leading to energy metabolism defects, impaired gut peristalsis, failure to empty the gut, and animal lethality [48]. The reduced AMPKα expression observed in NGLY1 deficiency cells was not caused by the loss of NFE2L1 activity. Restoration of Ngly1 or AMPKα expression could significantly alleviate the energy metabolism defects.
2.5. SLC12A2
Based on lethality association and co-evolution analysis, a conserved Na/K/Cl ion transporter Ncc69 (human NKCC1/2, officially SLC12A1/2) was identified, ubiquitously expressed and associated genetically with Ngly1 in Drosophila [48]. In Ncc69 or Ngly1 knockdown (KD) Drosophila, more than 30% showed severe seizures [49]. In mouse embryonic fibroblasts (MEFs), the homolog of Ncc69, NKCC1 was highly expressed and N-glycosylated, which was important for the function [50]. However, NGLY1 deficiency could disturb the N-glycosylation of NKCC1, which is detrimental to the functionality of the protein. In mammals, NKCC1 is expressed specifically in secretory epithelia, such as salivary, sweat, and lacrimal glands, to promote the basolateral ion absorption and subsequent secretion [51]. The decrease of NKCC1 functionality might account for the alacrima and reduced production of saliva and sweat observed in patients with NGLY1 deficiency.
The variety of functions carried out by this enzyme may explain the diversity and varying severity of symptoms caused by mutations in this gene.
3. Potential Treatments for NGLY1-CDDG
With the rapid increase of research in NGLY1’s structure, function and cellular pathways for cells and animal models in different species, several molecular methods were tested for the potential to treat NGLY1-deficient diseases.
3.1. Exogenous Restoration of NGLY1 Expression
Currently, double-knockout of NGLY1 has been used to study the enzymatic ability and molecular functions of the gene in several model organisms, such as yeast, Drosophila, worms, mouse and rat. After comparing the spectrum of clinical phenotypes and histological analysis, Ngly1−/− rats displayed very similar features to human NGLY1-deficiency patients [27]. Although Ngly1 was ubiquitously expressed, and highly transcribed in brain, lung, heart, kidney, liver, placenta and testis [52], the neurological symptoms in most patients implied the CNS as the most seriously affected organ [53].
AAV9s carrying a copy of human NGLY1 gene (AAV9-hNGLY1) were injected to Ngly1-KO SD rats during the weaning period via intra-cerebro-ventricular (i.c.v.) administration in order to reverse the deterioration of neuronal symptoms [54]. The re-expression of NGLY1 were identified in pons, thalamus, hippocampus, cerebral cortex and cerebellar Purkinje cells, but not detected in liver. The enzymatic activity of NGYL1 in Ngly1−/− rats was restored to a comparable level with that of wildtype controls. AAV9-hNGLY1 was safe and did not lead to liver toxicity. The motor dysfunction caused by NGLY1 deficiency, such as gait abnormalities, motor coordination and balance, were also significantly restored. However, the stride length and grip strength of limbs were not significantly ameliorated, which might be related to the absence of expression of functional NGLY1 in muscles. This was the first time that the expression of NGLY1 in a NGLY1-deficient mammalian model was restored. However, there are many questions to be answered: (1) Whether rat was suitable for studying the underlying molecular mechanisms of NGLY1-CDDG to explore potential drug targets. (2) Since NGLY1 was ubiquitously expressed, whether CNS was the most suitable organ to restore the exogenous NGLY1, and if the injection of AVV9-NGLY1 in more than one organ could ameliorate the clinical phenotypes to a higher degree. (3) Since primates were most closely related to humans in evolution, whether primates with NGLY1 deficiency might be the most appropriate animal model to study the pathogenesis of NGLY1-CDDG and to develop curable drugs.
3.2. Targeting ENGase with Small Inhibitors
It has been reported that endo-β-N-acetyl-glucosaminidase (ENGASE) could cleave the glycan moieties from N-linked glycoproteins at the beta-N-acetyl-glucosaminide, releasing a free Man5-9GlcNAc and proteins with a single GlcNAc residue linked to the Asn residual (N-GlcNAc proteins) (Figure 5C) [22]. The N-GlcNAc proteins are prone to aggregate, resulting in the dysfunction of the ERAD process. Additional deletion of ENGASE could partially prevent lethality and alleviate the phenotypes of the Ngly1-loss mice [55]. Therefore, inhibition of ENGASE might be a potential therapeutic site for NGLY1-deficient diseases. Currently, several novel ENGASE inhibitors have been discovered, including Proton Pump Inhibitors (PPIs), Lansoprazole, Rabeprazole and Omeprazole [56]. PPIs could immediately be considered as therapeutic drugs for treating NGLY1-deficiency because of their existing safety and pharmacokinetics profile.
3.3. Inhibition FOXB6 (Fbs2)
FOXB6 (also known as Fbs2) is a component of the SCF (SKP1-cullin-F-box) complex and functions as a substrate-binding adaptor (Figure 5B,C) [57]. In NGLY1-deficient cells, FOXB6 was overexpressed and resulted in cytotoxic impairment of the proteasome activity. The overexpressed FOXB6 not only induced ubiquitination of NFE2L1, but also inhibited NFE2L1’s processing by DDI2 and nuclear localizing in cells without NGLY1. Interestingly, additional knockout of Foxb6 could make Ngly1-deficient mice (Foxb6−/−; Ngly1−/−) viable and exhibit normal motor functions [58]. The survival ratio at P0 was two times higher than in Engase; Ngly1 dKO mice [55]. This indicates that FOXB6 might be a promising drug target for treatment of NGLY1-CDDG.
3.4. Activation of NFE2L2
It has been commonly accepted that inactivation of NFE2F1 was an important factor for the pathogenesis of NGLY1-CDDG. Functional loss of NFE2F1 could result in inhibition of proteasome function, mitochondrial dysfunction and immune dysregulation in NGLY1-deficient cells under proteotoxic stress [59]. In mammals, as a close homologue of NFE2F1, NFE2L2 (NRF2) was responsible for regulation of the expression of similar proteasome subunit genes as NFE2L1 under oxidative stress [60], and the expression of genes for autophagy and mitophagy [61][62]. Unlike the ER-bound NFE2FL1, NFE2L2 was cytosolic and non-N-glycosylated without relying on NGLY1 for its activation [63]. Increased expression of NFE2F2 could promote mitophagy and rescue the mitochondrial and immune homeostasis in Ngly1−/− cells. It has been verified that KEAP1 could strongly bind with and mediate the ubiquitination and degradation of NFE2L2 [64]. Therefore, chemical inhibitors to disrupt the KEAP1-NFE2L2 interaction might be a promising drug target. Interestingly, a natural inhibitor for KEAP1 derived from cruciferous vegetables (such as broccoli, cauliflower, kale, kohlrabi, Brussels sprouts and cabbage), sulforafane could efficiently bind with KEAP1 and robustly increase NFE2L2 protein level to promote the expression of proteasome subunit genes and mitophagy-related genes in NGLY1-deficient cells [59]. The disrupted mitochondria and dysregulated immune response to mtDNAs were significantly decreased after administration of sulforaphane, which indicated that KEAP1-NFE2L2 axis was a novel therapeutic site for correcting the abnormalities of NGLY1 diseases.
References
- Takahashi, N. Demonstration of a new amidase acting on glycopeptides. Biochem. Biophys. Res. Commun. 1977, 76, 1194–1201.
- Sugiyama, K.; Ishihara, H.; Tejima, S.; Takahashi, N. Demonstration of a new glycopeptidase, from jack-bean meal, acting on aspartylglucosylamine linkages. Biochem. Biophys. Res. Commun. 1983, 112, 155–160.
- Plummer, T.H., Jr.; Phelan, A.W.; Tarentino, A.L. Detection and quantification of peptide-N4-(N-acetyl-beta-glucosaminyl)asparagine amidases. Eur. J. Biochem. 1987, 163, 167–173.
- Diepold, A.; Li, G.; Lennarz, W.J.; Nurnberger, T.; Brunner, F. The Arabidopsis AtPNG1 gene encodes a peptide:N-glycanase. Plant J. Cell Mol. Biol. 2007, 52, 94–104.
- Takahashi, N.; Nishibe, H. Some characteristics of a new glycopeptidase acting on aspartylglycosylamine linkages. J. Biochem. 1978, 84, 1467–1473.
- Plummer, T.H., Jr.; Tarentino, A.L. Facile cleavage of complex oligosaccharides from glycopeptides by almond emulsin peptide:N-glycosidase. J. Biol. Chem. 1981, 256, 10243–10246.
- Nishibe, H.; Takahashi, N. The release of carbohydrate moieties from human fibrinogen by almond glycopeptidase without alteration in fibrinogen clottability. Biochim. Biophys. Acta 1981, 661, 274–279.
- Tarentino, A.L.; Plummer, T.H., Jr. Oligosaccharide accessibility to peptide:N-glycosidase as promoted by protein-unfolding reagents. J. Biol. Chem. 1982, 257, 10776–10780.
- Plummer, T.H., Jr.; Elder, J.H.; Alexander, S.; Phelan, A.W.; Tarentino, A.L. Demonstration of peptide:N-glycosidase F activity in endo-beta-N-acetylglucosaminidase F preparations. J. Biol. Chem. 1984, 259, 10700–10704.
- Ishii, K.; Iwasaki, M.; Inoue, S.; Kenny, P.T.; Komura, H.; Inoue, Y. Free sialooligosaccharides found in the unfertilized eggs of a freshwater trout, Plecoglossus altivelis. A large storage pool of complex-type bi-, tri-, and tetraantennary sialooligosaccharides. J. Biol. Chem. 1989, 264, 1623–1630.
- Seko, A.; Kitajima, K.; Iwasaki, M.; Inoue, S.; Inoue, Y. Structural studies of fertilization-associated carbohydrate-rich glycoproteins (hyosophorin) isolated from the fertilized and unfertilized eggs of flounder, Paralichthys olivaceus. Presence of a novel penta-antennary N-linked glycan chain in the tandem repeating glycopeptide unit of hyosophorin. J. Biol. Chem. 1989, 264, 15922–15929.
- Seko, A.; Kitajima, K.; Inoue, Y.; Inoue, S. Peptide:N-glycosidase activity found in the early embryos of Oryzias latipes (Medaka fish). The first demonstration of the occurrence of peptide:N-glycosidase in animal cells and its implication for the presence of a de-N-glycosylation system in living organisms. J. Biol. Chem. 1991, 266, 22110–22114.
- Suzuki, T.; Seko, A.; Kitajima, K.; Inoue, Y.; Inoue, S. Identification of peptide:N-glycanase activity in mammalian-derived cultured cells. Biochem. Biophys. Res. Commun. 1993, 194, 1124–1130.
- Ftouhi-Paquin, N.; Hauer, C.R.; Stack, R.F.; Tarentino, A.L.; Plummer, T.H., Jr. Molecular cloning, primary structure, and properties of a new glycoamidase from the fungus Aspergillus tubigensis. J. Biol. Chem. 1997, 272, 22960–22965.
- Suzuki, T.; Park, H.; Kitajima, K.; Lennarz, W.J. Peptides glycosylated in the endoplasmic reticulum of yeast are subsequently deglycosylated by a soluble peptide:N-glycanase activity. J. Biol. Chem. 1998, 273, 21526–21530.
- Need, A.C.; Shashi, V.; Hitomi, Y.; Schoch, K.; Shianna, K.V.; McDonald, M.T.; Meisler, M.H.; Goldstein, D.B. Clinical application of exome sequencing in undiagnosed genetic conditions. J. Med. Genet. 2012, 49, 353–361.
- Enns, G.M.; Shashi, V.; Bainbridge, M.; Gambello, M.J.; Zahir, F.R.; Bast, T.; Crimian, R.; Schoch, K.; Platt, J.; Cox, R.; et al. Mutations in NGLY1 cause an inherited disorder of the endoplasmic reticulum-associated degradation pathway. Genet. Med. 2014, 16, 751–758.
- Lam, C.; Ferreira, C.; Krasnewich, D.; Toro, C.; Latham, L.; Zein, W.M.; Lehky, T.; Brewer, C.; Baker, E.H.; Thurm, A.; et al. Prospective phenotyping of NGLY1-CDDG, the first congenital disorder of deglycosylation. Genet. Med. 2017, 19, 160–168.
- Haijes, H.A.; de Sain-van der Velden, M.G.M.; Prinsen, H.; Willems, A.P.; van der Ham, M.; Gerrits, J.; Couse, M.H.; Friedman, J.M.; van Karnebeek, C.D.M.; Selby, K.A.; et al. Aspartylglycosamine is a biomarker for NGLY1-CDDG, a congenital disorder of deglycosylation. Mol. Genet. Metab. 2019, 127, 368–372.
- Panneman, D.M.; Wortmann, S.B.; Haaxma, C.A.; van Hasselt, P.M.; Wolf, N.I.; Hendriks, Y.; Kusters, B.; van Emst-de Vries, S.; van de Westerlo, E.; Koopman, W.J.H.; et al. Variants in NGLY1 lead to intellectual disability, myoclonus epilepsy, sensorimotor axonal polyneuropathy and mitochondrial dysfunction. Clin. Genet. 2020, 97, 556–566.
- Hwang, J.; Qi, L. Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 2018, 43, 593–605.
- Hartl, F.U. Protein Misfolding Diseases. Annu. Rev. Biochem. 2017, 86, 21–26.
- Karousis, E.; Mühlemann, O. Nonsense-Mediated mRNA Decay Begins Where Translation Ends. Cold Spring Harb. Perspect. Biol. 2018, 11, a032862.
- Sovolyova, N.; Healy, S.; Samali, A.; Logue, S.E. Stressed to death—Mechanisms of ER stress-induced cell death. Biol. Chem. 2013, 395, 1–13.
- Kario, E.; Tirosh, B.; Ploegh, H.L.; Navon, A. N-Linked Glycosylation Does Not Impair Proteasomal Degradation but Affects Class I Major Histocompatibility Complex Presentation. J. Biol. Chem. 2008, 283, 244–254.
- Blom, D.; Hirsch, C.; Stern, P.; Tortorella, M.; Ploegh, H.L. A glycosylated type I membrane protein becomes cytosolic when peptide:N-glycanase is compromised. EMBO J. 2004, 23, 650–658.
- Owings, K.G.; Lowry, J.B.; Bi, Y.; Might, M.; Chow, C.Y. Transcriptome and functional analysis in a Drosophila model of NGLY1 deficiency provides insight into therapeutic approaches. Hum. Mol. Genet. 2018, 27, 1055–1066.
- Asahina, M.; Fujinawa, R.; Nakamura, S.; Yokoyama, K.; Tozawa, R.; Suzuki, T. Ngly1−/− rats develop neurodegenerative phenotypes and pathological abnormalities in their peripheral and central nervous systems. Hum. Mol. Genet. 2020, 29, 1635–1647.
- Tambe, M.A.; Ng, B.G.; Freeze, H.H. N-Glycanase 1 Transcriptionally Regulates Aquaporins Independent of Its Enzymatic Activity. Cell Rep. 2019, 29, 4620–4631.e4.
- Suzuki, T.; Park, H.; Hollingsworth, N.M.; Sternglanz, R.; Lennarz, W.J. PNG1, a Yeast Gene Encoding a Highly Conserved Peptide:N-Glycanase. J. Cell Biol. 2000, 149, 1039–1052.
- Mueller, W.F.; Jakob, P.; Sun, H.; Clauder-Münster, S.; Ghidelli-Disse, S.; Ordonez, D.; Boesche, M.; Bantscheff, M.; Collier, P.; Haase, B.; et al. Loss of N-Glycanase 1 Alters Transcriptional and Translational Regulation in K562 Cell Lines. G3 Genes Genomes Genet. 2020, 10, 1585–1597.
- Misaghi, S.; Pacold, M.E.; Blom, D.; Ploegh, H.L.; Korbel, G.A. Using a Small Molecule Inhibitor of Peptide:N-Glycanase to Probe Its Role in Glycoprotein Turnover. Chem. Biol. 2004, 11, 1677–1687.
- Kim, H.M.; Han, J.W.; Chan, J.Y. Nuclear Factor Erythroid-2 Like 1 (NFE2L1): Structure, function and regulation. Gene 2016, 584, 17–25.
- Steffen, J.; Seeger, M.; Koch, A.; Krüger, E. Proteasomal Degradation Is Transcriptionally Controlled by TCF11 via an ERAD-Dependent Feedback Loop. Mol. Cell 2010, 40, 147–158.
- Lehrbach, N.; Breen, P.C.; Ruvkun, G. Protein Sequence Editing of SKN-1A/Nrf1 by Peptide:N-Glycanase Controls Proteasome Gene Expression. Cell 2019, 177, 737–750.e15.
- Lehrbach, N.; Ruvkun, G. Proteasome dysfunction triggers activation of SKN-1A/Nrf1 by the aspartic protease DDI-1. eLife 2016, 5, e17721.
- Koizumi, S.; Irie, T.; Hirayama, S.; Sakurai, Y.; Yashiroda, H.; Naguro, I.; Ichijo, H.; Hamazaki, J.; Murata, S. The aspartyl protease DDI2 activates Nrf1 to compensate for proteasome dysfunction. eLife 2016, 5, e18357.
- Murphy, P.; Kolstø, A.-B. Expression of the bZIP transcription factor TCF11 and its potential dimerization partners during development. Mech. Dev. 2000, 97, 141–148.
- Delporte, C.; Bryla, A.; Perret, J. Aquaporins in Salivary Glands: From Basic Research to Clinical Applications. Int. J. Mol. Sci. 2016, 17, 166.
- Zhi, H.; Yuan, W.-T. Expression of aquaporin 3, 4, and 8 in colonic mucosa of rat models with slow transit constipation. Zhonghua Wei Chang Wai Ke Za Zhi = Chin. J. Gastrointest. Surg. 2011, 14, 1124–1130.
- Galeone, A.; Han, S.Y.; Huang, C.; Hosomi, A.; Suzuki, T.; Jafar-Nejad, H. Tissue-specific regulation of BMP signaling by Drosophila N-glycanase 1. eLife 2017, 6, e27612.
- Galeone, A.; Adams, J.; Matsuda, S.; Presa, M.F.; Pandey, A.; Han, S.Y.; Tachida, Y.; Hirayama, H.; Vaccari, T.; Suzuki, T.; et al. Regulation of BMP4/Dpp retrotranslocation and signaling by deglycosylation. eLife 2020, 9, e55596.
- Bakrania, P.; Efthymiou, M.; Klein, J.C.; Salt, A.; Bunyan, D.J.; Wyatt, A.; Ponting, C.P.; Martin, A.; Williams, S.; Lindley, V.; et al. Mutations in BMP4 Cause Eye, Brain, and Digit Developmental Anomalies: Overlap between the BMP4 and Hedgehog Signaling Pathways. Am. J. Hum. Genet. 2008, 82, 304–319.
- Obara-Ishihara, T.; Kuhlman, J.; Niswander, L.; Herzlinger, D. The surface ectoderm is essential for nephric duct formation in intermediate mesoderm. Development 1999, 126, 1103–1108.
- Tirosh-Finkel, L.; Zeisel, A.; Brodt-Ivenshitz, M.; Shamai, A.; Yao, Z.; Seger, R.; Domany, E.; Tzahor, E. BMP-mediated inhibition of FGF signaling promotes cardiomyocyte differentiation of anterior heart field progenitors. Development 2010, 137, 2989–3000.
- Reis, L.M.; Tyler, R.C.; Schilter, K.F.; Abdul-Rahman, O.; Innis, J.W.; Kozel, B.A.; Schneider, A.S.; Bardakjian, T.M.; Lose, E.J.; Martin, D.M.; et al. BMP4 loss-of-function mutations in developmental eye disorders including SHORT syndrome. Qual. Life Res. 2011, 130, 495–504.
- Kong, J.; Peng, M.; Ostrovsky, J.; Kwon, Y.J.; Oretsky, O.; McCormick, E.M.; He, M.; Argon, Y.; Falk, M.J. Mitochondrial function requires NGLY1. Mitochondrion 2017, 38, 6–16.
- Han, S.Y.; Pandey, A.; Moore, T.; Galeone, A.; Duraine, L.; Cowan, T.M.; Jafar-Nejad, H. A conserved role for AMP-activated protein kinase in NGLY1 deficiency. PLoS Genet. 2020, 16, e1009258.
- Talsness, D.M.; Owings, K.G.; Coelho, E.; Mercenne, G.; Pleinis, J.M.; Partha, R.; A Hope, K.; Zuberi, A.R.; Clark, N.L.; Lutz, C.M.; et al. A Drosophila screen identifies NKCC1 as a modifier of NGLY1 deficiency. eLife 2020, 9, e27831.
- Rusan, Z.M.; Kingsford, O.A.; Tanouye, M.A. Modeling Glial Contributions to Seizures and Epileptogenesis: Cation-Chloride Cotransporters in Drosophila melanogaster. PLoS ONE 2014, 9, e101117.
- Singh, R.; Almutairi, M.M.; Pacheco-Andrade, R.; Almiahuob, M.Y.M.; Di Fulvio, M. Impact of Hybrid and Complex N-Glycans on Cell Surface Targeting of the Endogenous Chloride Cotransporter Slc12a2. Int. J. Cell Biol. 2015, 2015, 505294.
- Lipiński, P.; Bogdańska, A.; Różdżyńska-Świątkowska, A.; Wierzbicka-Rucińska, A.; Tylki-Szymańska, A. NGLY1 deficiency: Novel patient, review of the literature and diagnostic algorithm. JIMD Rep. 2020, 51, 82–88.
- Abuduxikuer, K.; Zou, L.; Wang, L.; Chen, L.; Wang, J.-S. Novel NGLY1 gene variants in Chinese children with global developmental delay, microcephaly, hypotonia, hypertransaminasemia, alacrimia, and feeding difficulty. J. Hum. Genet. 2020, 65, 387–396.
- Suzuki, T.; A Kwofie, M.; Lennarz, W.J. Ngly1, a mouse gene encoding a deglycosylating enzyme implicated in proteasomal degradation: Expression, genomic organization, and chromosomal mapping. Biochem. Biophys. Res. Commun. 2003, 304, 326–332.
- Asahina, M.; Fujinawa, R.; Hirayama, H.; Tozawa, R.; Kajii, Y.; Suzuki, T. Reversibility of motor dysfunction in the rat model of NGLY1 deficiency. Mol. Brain 2021, 14, 91.
- Fujihira, H.; Masahara-Negishi, Y.; Tamura, M.; Huang, C.; Harada, Y.; Wakana, S.; Takakura, D.; Kawasaki, N.; Taniguchi, N.; Kondoh, G.; et al. Lethality of mice bearing a knockout of the Ngly1-gene is partially rescued by the additional deletion of the Engase gene. PLoS Genet. 2017, 13, e1006696.
- Yoshida, Y.; Chiba, T.; Tokunaga, F.; Kawasaki, H.; Iwai, K.; Suzuki, T.; Ito, Y.; Matsuoka, K.; Yoshida, M.; Tanaka, K.; et al. E3 ubiquitin ligase that recognizes sugar chains. Nature 2002, 418, 438–442.
- Bi, Y.; Might, M.; Vankayalapati, H.; Kuberan, B. Repurposing of Proton Pump Inhibitors as first identified small molecule inhibitors of endo -β- N -acetylglucosaminidase (ENGase) for the treatment of NGLY1 deficiency, a rare genetic disease. Bioorganic Med. Chem. Lett. 2017, 27, 2962–2966.
- Yoshida, Y.; Asahina, M.; Murakami, A.; Kawawaki, J.; Yoshida, M.; Fujinawa, R.; Iwai, K.; Tozawa, R.; Matsuda, N.; Tanaka, K.; et al. Loss of peptide:N-glycanase causes proteasome dysfunction mediated by a sugar-recognizing ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2021, 118, e2102922118.
- Yang, K.; Huang, R.; Fujihira, H.; Suzuki, T.; Yan, N. N-glycanase NGLY1 regulates mitochondrial homeostasis and inflammation through NRF1. J. Exp. Med. 2018, 215, 2600–2616.
- Kwak, M.-K.; Wakabayashi, N.; Greenlaw, J.L.; Yamamoto, M.; Kensler, T.W. Antioxidants Enhance Mammalian Proteasome Expression through the Keap1-Nrf2 Signaling Pathway. Mol. Cell. Biol. 2003, 23, 8786–8794.
- East, D.A.; Fagiani, F.; Crosby, J.; Georgakopoulos, N.D.; Bertrand, H.; Schaap, M.; Fowkes, A.; Wells, G.; Campanella, M. PMI: A ΔΨm Independent Pharmacological Regulator of Mitophagy. Chem. Biol. 2014, 21, 1585–1596.
- Pajares, M.; Jiménez-Moreno, N.; García-Yagüe, J.; Escoll, M.; de Ceballos, M.L.; Van Leuven, F.; Rábano, A.; Yamamoto, M.; Rojo, A.I.; Cuadrado, A. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 2016, 12, 1902–1916.
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell Survival Responses to Environmental Stresses Via the Keap1-Nrf2-ARE Pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
822
Revisions:
3 times
(View History)
Update Date:
20 Jun 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No