 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Bikash Pattnaik | + 1300 word(s) | 1300 | 2020-10-07 04:18:54 | | | |
| 2 | Catherine Yang | Meta information modification | 1300 | 2020-10-10 12:01:57 | | |
Video Upload Options
Ionchannels are membrane protein that allows the flow of ions across the cell membrane. This is essential for cellular communication and hence mutations that results in non functional protein cause disease. The paper reviews only nonsense mutations of ion channels that cause blindness and proposes several ways this can be corrected.
1. Introduction
Ion channels are membrane-spanning transport proteins that facilitate the passive bidirectional (into and out of cell and cell organelles) flow of selective ions like sodium (Na+), potassium (K+), calcium (Ca2+), chloride (Cl−), or unspecific cations (Figure 1). These ion channel proteins have an enormous heterogeneity in their electrophysiological properties. In the eye, numerous ion channels are found at different anatomical locations like cornea, lens, ciliary body, and retina, as outlined in Figure 2. A flux of ions generates a membrane potential that maintains the ionic homeostasis required for the proper functioning of cells like signal transmission and visual processing in ocular cells [1][2][3][4]. Ion channels are either gated or non-gated, which permit selective ions to pass through the membrane based on the electrochemical gradient. Gated ion channels are regulated by electrical (membrane potential, voltage), chemical (ligands, cyclic nucleotides, phosphorylation), or other signals (light, temperature, pH, or mechanical stimuli). In contrast, non-gated channels allow the free flow of ions across the membrane.
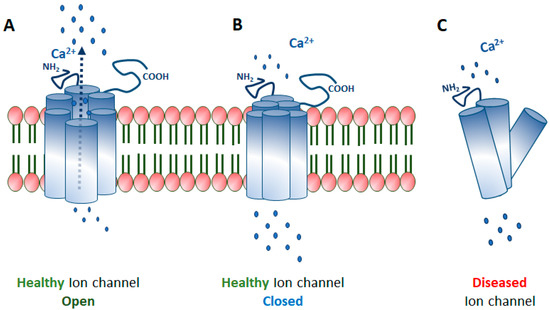
Figure 1. Ion channels; (A) An ion channel is open, allowing the flow of Ca2+ ions, (B) A closed ion channel limiting the flow of ions, (C) A truncated (diseased) ion channel not trafficked to the membrane due to dismantled subunits.
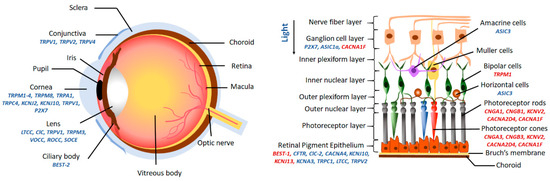
Figure 2. Anatomical location of ocular ion channels. Channels highlighted in the red cause associated blindness due to mutations.
The steady-state regulation of ionic balance by channel proteins is a critical phenomenon in the eye for most of the cellular functions. The most common is the cyclic nucleotide-gated (CNG) cationic channel, which mediates phototransduction in rod and cone photoreceptors by regulating ligand-dependent homeostasis (Na+ and Ca2+) [5][6][7][8][9]. Ca2+ homeostasis is also required to regulate the physiology of the lens, and an overload of Ca2+ is detrimental to the lens, leading to cortical cataract [10]. Different combinations of channels (voltage-operated Ca2+ channels; VOCCs, receptor-operated Ca2+ channels; ROCCs, second messenger-operated Ca2+ channels; SOCE, transient receptor potential channels; TRPs) tightly regulate the Ca2+ influx in lens to maintain its transparency [10][11]. Voltage and ligand operated Ca2+, K+, Cl− channels in the retinal pigmented epithelial (RPE) layer contribute to the secretory activity, volume regulation, and transepithelial ion transport to maintain the retinal health. In contrast, alterations in these channel functions lead to retinal degeneration [12]. K+ channel activity modulates essential action potential in excitable cells and transport and secretory activity in non-excitable cells. K+ channels (inwardly rectifying K+ (Kir)-channels, voltage-gated K+ channels (Kv), Ca2+-activated K+ channels, two-pore or leak K+-channels) are imperative in ocular tissues like the cornea to regulate epithelial cell proliferation and apoptosis [13][14][15], the lens to maintain the volume and transparency [16], the retinal ganglion cells to modulate the resting membrane potential and cell excitability [17], and the RPE cell physiology, for its interaction with the photoreceptor and ionic composition of the subretinal space [18][19]. The passive flow of Cl− in ocular tissues like cornea, conjunctiva, ciliary epithelium, and RPE is mediated by Cl− channels (high- (maxi) conductance, the cystic fibrosis transmembrane conductance regulator (CFTR), volume-regulated, voltage-gated, Ca2+-activated) to regulate tear film volume, aqueous humor volume, corneal transparency, and ionic composition [20][21][22][23][24][25][26][27][28][29][30].
The human genome has over 400 ion channel genes, and several mutations have been reported across several ion channel genes leading to various pathological conditions, known as channelopathies [31]. These mutations may alter the structure, assembly, localization, trafficking, or functions of channel protein and, therefore, turn a sensing ion channel into a non-sensing one (Figure 1C). The effect of these mutations can be studied by electrophysiological techniques. Gene mutations can result in a loss or gain of functions. Structurally, these channel proteins are formed by the assembly of several similar (homomeric) or different (heteromeric) subunits. Therefore, loss-of-function mutations sometimes result in a dominant-negative effect in ion channels. Most of the ocular channelopathies are rare conditions and have been a rapidly expanding, yet unexplored area of ophthalmology. There is a substantial amount of genetic and allelic heterogeneity in channelopathies. It remains unclear how different mutations in the same gene can result in a wide range of phenotypic variability. For example, BEST1 gene mutations result in a spectrum of ocular phenotype such as microcornea, cataract, retinitis pigmentosa, and macular and rod-cone dystrophy [32]. Interestingly, mutations in the same gene exhibit a different inheritance pattern. For example, mutations in the KCNJ13 gene lead to LCA16, an autosomal recessive disease, and snowflake vitreoretinal degeneration (SVD), an autosomal dominant disease [33][34][35].
Many ion channel genes are expressed in the eye, and few are known to harbor the disease-causing mutations. These genes involved in ocular ion channelopathies were identified by chromosomal mapping and targeted exome sequencing (KCNJ13; [35]), microsatellite analysis (CACNA1F; [36]), polymorphism analysis, and recombination mapping (BEST1; [37]), based on the phenotype in a mutant mouse model (CACNA2D4; [38]) and, homozygosity mapping and linkage analysis (KCNV2 [39], CNGA3 [40], CNGB3 [41], CNGB1 [41], CNGA1 [42], TRPM1 [43]). The present article reviews the fundamentals of ocular ion channelopathies caused by these genes, with an exclusive insight into the role of genetic mutations in disease pathogenesis. We also discuss the future percepts of potential pharmacological and therapeutic strategies.
2. Potential Therapies for Reshaping the Non-Sensing Ocular Ion Channels to Sensing Ones
Genetic mutations contribute significantly to the stunning array of ocular channelopathies. The united efforts of clinicians and scientists enabled the discovery of the association of different ion channel genes to the disease phenotype. With the advances in technology, screening mutations across these genes and their effect on specific channel protein function have become possible. It is now known that these ion channels are multimeric in nature and function in-network with several other regulatory proteins. Research on the biophysical properties of ion channels has revealed the fundamental processes responsible for the selective nature of these proteins to understand the normal physiology. These details will help scientists provide a new promising paradigm for precise diagnosis of disease to facilitate the development of rational treatments. Several therapies (Figure 3) and clinical trials targeting the disease-specific genes (mutations), transcripts, or proteins in various ocular tissues are now in practice (Table 1). One advantage of testing the potential therapies in the eye is its easy accessibility for surgical intervention, non-invasive optical imaging, monitoring, and delivering different therapeutics. Also, the presence of the blood-ocular barrier makes the eye immune-privileged and limits the toxicity of drugs and therapies. Innovating the novel therapeutic approaches and their efficient delivery to the eye for various channelopathies is a viable treatment option.
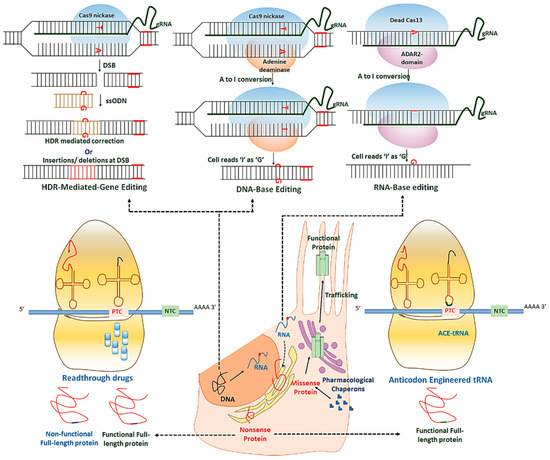
Figure 3. Potential therapies for ocular channelopathies caused by gene mutations.
Table 1. Therapies for ion channelopathies in practice and clinical trials.
| Gene | Therapy In Vitro/In Vivo | References |
|---|---|---|
| KCNJ13 | AAV gene therapy in iPSC-RPE in vitro | [44] |
| Read through in iPSC-RPE in vitro | [44] | |
| TRPM1 | AAV gene therapy in mice | [45] |
| BEST1 | AAV2-gene therapy in dogs | [46] |
| Gene augmentation and CRISPR-gene editing in iPSC-RPE | [47] | |
| Pharmacological chaperons in culture cells (MDCK/HEK293) | [48] | |
| CNGA3 | AAV5-mediated gene therapy in mice AAV5-mediated gene augmentation in sheep |
[49][50] |
| NGB3 | AAV-gene therapy in mice | [51][52] |
| CNGB1 | AAV-gene therapy in mice | [53][54] |
3. Concluding Remarks
Ocular ion channel proteins are essential for visual functions and processing. Although many important questions regarding stoichiometry and assembly of subunits along with biophysical and physiological functions of ion channel proteins have been answered, further studies are needed to address the impact of mutations in disease mechanism and pathophysiology to formulate the specific therapies. It is essential to ultimately translate all laboratory research-based therapies to clinics for the treatment of patients who have blindness caused by channelopathies. A combinational therapy or a cocktail of different genetic and/or pharmacological therapeutic agents at a nontoxic level could be a key for reversing the nonsense ion-channels to sense and restore vision.
References
- Krizaj, D.; Copenhagen, D.R. Calcium regulation in photoreceptors. Front. Biosci. 2002, 7, d2023–d2044.
- Heidelberger, R.; Thoreson, W.B.; Witkovsky, P. Synaptic transmission at retinal ribbon synapses. Prog. Retin. Eye Res. 2005, 24, 682–720.
- Nelson, R.; Connaughton, V. Bipolar Cell Pathways in the Vertebrate Retina; University of Utah Health Sciences Center: Salt Lake City, UT, USA, 1995.
- Protti, D.A.; Llano, I. Calcium Currents and Calcium Signaling in Rod Bipolar Cells of Rat Retinal Slices. J. Neurosci. 1998, 18, 3715–3724.
- Fesenko, E.E.; Kolesnikov, S.S.; Lyubarsky, A.L. Induction by cyclic GMP of cationic conductance in plasma membrane of retinal rod outer segment. Nature 1985, 313, 310–313.
- Yau, K.-W.; Nakatani, K. Light-suppressible, cyclic GMP-sensitive conductance in the plasma membrane of a truncated rod outer segment. Nature 1985, 317, 252–255.
- Haynes, L.W.; Kay, A.R.; Yau, K.-W. Single cyclic GMP-activated channel activity in excised patches of rod outer segment membrane. Nature 1986, 321, 66–70.
- Tanaka, J.C.; Furman, R.E.; Cobbs, W.H.; Mueller, P. Incorporation of a retinal rod cGMP-dependent conductance into planar bilayers. Proc. Natl. Acad. Sci. USA 1987, 84, 724–728.
- Haynes, L.W.; Yau, K.W. Single-channel measurement from the cyclic GMP-activated conductance of catfish retinal cones. J. Physiol. 1990, 429, 451–481.
- Rhodes, J.D.; Sanderson, J. The mechanisms of calcium homeostasis and signalling in the lens. Exp. Eye Res. 2009, 88, 226–234.
- Maddala, R.; Nagendran, T.; De Ridder, G.G.; Schey, K.L.; Rao, P.V. L-Type Calcium Channels Play a Critical Role in Maintaining Lens Transparency by Regulating Phosphorylation of Aquaporin-0 and Myosin Light Chain and Expression of Connexins. PLoS ONE 2013, 8, e64676.
- Wimmers, S.; Karl, M.O.; Strauß, O. Ion channels in the RPE. Prog. Retin. Eye Res. 2007, 26, 263–301.
- Candia, O.A.; Zamudio, A. Cl Secretagogues Reduce Basolateral K Permeability in the Rabbit Corneal Epithelium. J. Membr. Biol. 2002, 190, 197–205.
- Roderick, C.; Reinach, P.S.; Wang, L.; Lu, L. Modulation of Rabbit Corneal Epithelial Cell Proliferation by Growth Factor-regulated K+ Channel Activity. J. Membr. Biol. 2003, 196, 41–50.
- Wolosin, J.M.; Candia, O.A. Cl− secretagogues increase basolateral K+ conductance of frog corneal epithelium. Am. J. Physiol. Physiol. 1987, 253, C555–C560.
- Cooper, K.; Rae, J.L.; Dewey, J. Inwardly rectifying potassium current in mammalian lens epithelial cells. Am. J. Physiol. Physiol. 1991, 261, C115–C123.
- Zhong, Y.; Wang, J.; Liu, W.; Zhu, Y. Potassium ion channels in retinal ganglion cells (Review). Mol. Med. Rep. 2013, 8, 311–319.
- Kumar, M.; Pattnaik, B.R. Focus on Kir7.1: Physiology and channelopathy. Channels 2014, 8, 488–495.
- Reichhart, N.; Strauß, O. Ion channels and transporters of the retinal pigment epithelium. Exp. Eye Res. 2014, 126, 27–37.
- Levin, M.H.; Verkman, A.S. CFTR-Regulated Chloride Transport at the Ocular Surface in Living Mice Measured by Potential Differences. Investig. Opthalmol. Vis. Sci. 2005, 46, 1428–1434.
- Levin, M.; Verkman, A.S. Aquaporins and CFTR in Ocular Epithelial Fluid Transport. J. Membr. Biol. 2006, 210, 105–115.
- Edelman, J.L.; Loo, D.D.; Sachs, G. Characterization of potassium and chloride channels in the basolateral membrane of bovine nonpigmented ciliary epithelial cells. Investig. Ophthalmol. Vis. Sci. 1995, 36, 2706–2716.
- Zhang, H.; Wong, C.L.; Shan, S.W.; Li, K.K.; Cheng, A.K.; Lee, K.-L.D.; Ge, J.; To, C.-H.; Do, C. Characterisation of Cl− transporter and channels in experimentally induced myopic chick eyes. Clin. Exp. Optom. 2011, 94, 528–535.
- Shiue, M.H.I.; Gukasyan, H.J.; Kim, K.; Loo, D.D.F.; Lee, V.H.L. Characterization of cyclic AMP-regulated chloride conductance in the pigmented rabbit conjunctival epithelial cells. Can. J. Physiol. Pharm. 2002, 80, 533–540.
- Turner, H.C.; Bernstein, A.; Candia, O.A. Presence of CFTR in the conjunctival epithelium. Curr. Eye Res. 2002, 24, 182–187.
- Sun, X.C.; Bonanno, J.A. Expression, localization, and functional evaluation of CFTR in bovine corneal endothelial cells. Am. J. Physiol. Physiol. 2002, 282, C673–C683.
- Chen, S.; Mead, A.; Suzuki, Y.; Sears, M. Basolateral membrane chloride channels in rabbit non-pigmented ciliary epithelium. Investig. Ophthalmol. Vis. Sci. 1996, 37, 670.
- Do, C.; Peterson-Yantorno, K.; Mitchell, C.H.; Civan, M.M. cAMP-activated maxi-Cl− channels in native bovine pigmented ciliary epithelial cells. Am. J. Physiol. Physiol. 2004, 287, C1003–C1011.
- Reigada, D.; Mitchell, C.H. Release of ATP from retinal pigment epithelial cells involves both CFTR and vesicular transport. Am. J. Physiol. Physiol. 2005, 288, C132–C140.
- Jacob, T.J.C.; Zhang, J.J. Chloride channels in bovine pigmented and non-pigmented ciliary epithelial cells. Investig. Ophthalmol. Vis. Sci. 1996, 37, 2011.
- Wilde, A.A.; Amin, A. Channelopathies, genetic testing and risk stratification. Int. J. Cardiol. 2017, 237, 53–55.
- Boon, C.J.; Klevering, B.J.; Leroy, B.P.; Hoyng, C.B.; Keunen, J.E.; Hollander, A.I.D. The spectrum of ocular phenotypes caused by mutations in the BEST1 gene. Prog. Retin. Eye Res. 2009, 28, 187–205.
- Pattnaik, B.R.; Shahi, P.K.; Marino, M.J.; Liu, X.; York, N.; Brar, S.; Chiang, J.; Pillers, D.-A.M.; Traboulsi, E.I. A NovelKCNJ13Nonsense Mutation and Loss of Kir7.1 Channel Function Causes Leber Congenital Amaurosis (LCA16). Hum. Mutat. 2015, 36, 720–727.
- Hejtmancik, J.F.; Jiao, X.; Li, A.; Sergeev, Y.V.; Ding, X.; Sharma, A.K.; Chan, C.-C.; Medina, I.; Edwards, A.O. Mutations in KCNJ13 Cause Autosomal-Dominant Snowflake Vitreoretinal Degeneration. Expand. Spectr. Baf-Relat. Disord. Novo Var. Smarcc2 Cause Syndr. Intellect. Disabil. Dev. Delay 2008, 82, 174–180.
- Sergouniotis, P.I.; Davidson, A.E.; Mackay, D.S.; Li, Z.; Yang, X.; Plagnol, V.; Moore, A.T.; Webster, A.R. Recessive Mutations in KCNJ13, Encoding an Inwardly Rectifying Potassium Channel Subunit, Cause Leber Congenital Amaurosis. Am. J. Hum. Genet. 2011, 89, 183–190.
- Strom, T.M.; Nyakatura, G.; Apfelstedt-Sylla, E.; Hellebrand, H.; Lorenz, B.; Weber, B.H.F.; Wutz, K.; Gutwillinger, N.; Rüther, K.; Drescher, B.; et al. An L-type calcium-channel gene mutated in incomplete X-linked congenital stationary night blindness. Nat. Genet. 1998, 19, 260–263.
- Petrukhin, K.; Koisti, M.J.; Bakall, B.; Li, W.; Xie, G.; Marknell, T.; Sandgren, O.; Forsman, K.; Holmgren, G.; Andreasson, S.; et al. Identification of the gene responsible for Best macular dystrophy. Nat. Genet. 1998, 19, 241–247.
- Wycisk, K.A.; Budde, B.; Feil, S.; Skosyrski, S.; Buzzi, F.; Neidhardt, J.; Glaus, E.; Nürnberg, P.; Ruether, K.; Berger, W. Structural and Functional Abnormalities of Retinal Ribbon Synapses due toCacna2d4Mutation. Investig. Opthalmol. Vis. Sci. 2006, 47, 3523–3530.
- Wu, H.; Cowing, J.A.; Michaelides, M.; Wilkie, S.E.; Jeffery, G.; Jenkins, S.A.; Mester, V.; Bird, A.C.; Robson, A.G.; Holder, G.E.; et al. Mutations in the Gene KCNV2 Encoding a Voltage-Gated Potassium Channel Subunit Cause “Cone Dystrophy with Supernormal Rod Electroretinogram” in Humans. Expand. Spectr. Baf-Relat. Disord. Novo Var. Smarcc2 Cause Syndr. Intellect. Disabil. Dev. Delay 2006, 79, 574–579.
- Hussels, I.E.; Morton, N.E. Pingelap and Mokil Atolls: Achromatopsia. Am. J. Hum. Genet. 1972, 24, 304–309.
- Sundin, O.H.; Yang, J.-M.; Li, Y.; Zhu, D.; Hurd, J.N.; Mitchell, T.N.; Silva, E.D.; Maumenee, I.H. Genetic basis of total colourblindness among the Pingelapese islanders. Nat. Genet. 2000, 25, 289–293.
- Dryja, T.P.; Finn, J.T.; Peng, Y.W.; McGee, T.L.; Berson, E.L.; Yau, K.W. Mutations in the gene encoding the alpha subunit of the rod cGMP-gated channel in autosomal recessive retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 1995, 92, 10177–10181.
- Li, Z.; Sergouniotis, P.I.; Michaelides, M.; Mackay, D.S.; Wright, G.A.; Devery, S.; Moore, A.T.; Holder, G.E.; Robson, A.G.; Webster, A.R. Recessive Mutations of the Gene TRPM1 Abrogate ON Bipolar Cell Function and Cause Complete Congenital Stationary Night Blindness in Humans. Am. J. Hum. Genet. 2009, 85, 711–719.
- Shahi, P.K.; Hermans, D.; Sinha, D.; Brar, S.; Moulton, H.; Stulo, S.; Borys, K.D.; Capowski, E.; Pillers, D.-A.M.; Gamm, D.; et al. Gene Augmentation and Readthrough Rescue Channelopathy in an iPSC-RPE Model of Congenital Blindness. Am. J. Hum. Genet. 2019, 104, 310–318.
- Scalabrino, M.L.; Boye, S.L.; Fransen, K.M.H.; Noel, J.M.; Dyka, F.M.; Min, S.H.; Ruan, Q.; De Leeuw, C.N.; Simpson, E.M.; Gregg, R.G.; et al. Intravitreal delivery of a novel AAV vector targets ON bipolar cells and restores visual function in a mouse model of complete congenital stationary night blindness. Hum. Mol. Genet. 2015, 24, 6229–6239.
- Guziewicz, K.E.; Cideciyan, A.V.; Beltran, W.A.; Komaromy, A.M.; Dufour, V.; Swider, M.; Iwabe, S.; Sumaroka, A.; Kendrick, B.T.; Ruthel, G.; et al. BEST1 gene therapy corrects a diffuse retina-wide microdetachment modulated by light exposure. Proc. Natl. Acad. Sci. USA 2018, 115, E2839–E2848.
- Sinha, D.; Steyer, B.G.; Shahi, P.K.; Mueller, K.; Valiauga, R.; Edwards, K.L.; Bacig, C.; Steltzer, S.S.; Srinivasan, S.; Abdeen, A.; et al. Human iPSC modeling reveals mutation-specific responses to gene therapy in Best disease. bioRxiv 2020, 796581.
- Uggenti, C.; Briant, K.; Streit, A.-K.; Thomson, S.; Koay, Y.H.; Baines, R.A.; Swanton, E.; Manson, F.D. Restoration of mutant bestrophin-1 expression, localisation and function in a polarised epithelial cell model. Dis. Model. Mech. 2016, 9, 1317–1328.
- Michalakis, S.; Mühlfriedel, R.; Tanimoto, N.; Krishnamoorthy, V.; Koch, S.; Fischer, M.D.; Becirovic, E.; Bai, L.; Huber, G.; Beck, S.C.; et al. Restoration of Cone Vision in the CNGA3−/−Mouse Model of Congenital Complete Lack of Cone Photoreceptor Function. Mol. Ther. 2010, 18, 2057–2063.
- Banin, E.; Gootwine, E.; Obolensky, A.; Ezra-Elia, R.; Ejzenberg, A.; Zelinger, L.; Honig, H.; Rosov, A.; Yamin, E.; Sharon, O.; et al. Gene Augmentation Therapy Restores Retinal Function and Visual Behavior in a Sheep Model of CNGA3 Achromatopsia. Mol. Ther. 2015, 23, 1423–1433.
- Carvalho, L.S.; Xu, J.; Pearson, R.A.; Smith, A.J.; Bainbridge, J.W.; Morris, L.M.; Fliesler, S.J.; Ding, X.-Q.; Ali, R.R. Long-term and age-dependent restoration of visual function in a mouse model of CNGB3-associated achromatopsia following gene therapy. Hum. Mol. Genet. 2011, 20, 3161–3175.
- Pang, J.-J.; Deng, W.-T.; Dai, X.; Lei, B.; Everhart, E.; Umino, Y.; Li, J.; Zhang, K.; Mao, S.; Boye, S.L.; et al. AAV-Mediated Cone Rescue in a Naturally Occurring Mouse Model of CNGA3-Achromatopsia. PLoS ONE 2012, 7, e35250.
- Koch, S.; Sothilingam, V.; Garrido, M.G.; Tanimoto, N.; Becirovic, E.; Seide, C.; Beck, S.C.; Seeliger, M.W.; Biel, M.; Mühlfriedel, R.; et al. Gene therapy restores vision and delays degeneration in the CNGB1−/−mouse model of retinitis pigmentosa. Hum. Mol. Genet. 2012, 21, 4486–4496.
- Michalakis, S.; Koch, S.; Sothilingam, V.; Garrido, M.G.; Tanimoto, N.; Schulze, E.; Becirovic, E.; Koch, F.; Seide, C.; Beck, S.C.; et al. Gene Therapy Restores Vision and Delays Degeneration in the CNGB1−/−Mouse Model of Retinitis Pigmentosa. Adv. Exp. Med. Biol. 2014, 801, 733–739.

