Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Pavel Kopnin | -- | 2171 | 2022-06-16 08:15:15 | | | |
| 2 | Sirius Huang | Meta information modification | 2171 | 2022-06-16 09:08:53 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Kopnin, P.; , .; Kudlay, D. Senescent Fibroblasts and Skin Aging. Encyclopedia. Available online: https://encyclopedia.pub/entry/24095 (accessed on 24 June 2026).
Kopnin P, , Kudlay D. Senescent Fibroblasts and Skin Aging. Encyclopedia. Available at: https://encyclopedia.pub/entry/24095. Accessed June 24, 2026.
Kopnin, Pavel, , Dmitry Kudlay. "Senescent Fibroblasts and Skin Aging" Encyclopedia, https://encyclopedia.pub/entry/24095 (accessed June 24, 2026).
Kopnin, P., , ., & Kudlay, D. (2022, June 16). Senescent Fibroblasts and Skin Aging. In Encyclopedia. https://encyclopedia.pub/entry/24095
Kopnin, Pavel, et al. "Senescent Fibroblasts and Skin Aging." Encyclopedia. Web. 16 June, 2022.
Copy Citation
Skin aging is a multi-factorial process that affects nearly every aspect of skin biology and function. The processes developing in the skin during aging are based on fundamental molecular mechanisms associated with fibroblasts, the main cellular population of the dermis. It has been revealed that the amount of fibroblasts decreases markedly with age and their functional activity is also reduced. This inevitably leads to a decrease in the regenerative abilities of the skin and the progression of its aging.
skin
aging
fibroblasts
stem cells
1. Introduction
Senescent dermal fibroblasts (senDFs) like other senescent cells (senCs) undergo morphological changes, promoting their resistance to apoptosis, which contributes to the accumulation of these cells in the dermis [1][2] and changes the pattern of gene expression. The resistance of senDFs to apoptosis may be caused by an increase in the levels of anti-apoptotic proteins such as BCL-2 and FOXO4 [3][4]. It may also be associated with the secretion of non-classical MHC HLA-E, which activates inhibitory receptor NKG2A on natural killer cells (NK cells) and CD8+ T lymphocytes. Activation of NKG2A prevents the elimination of senDFs [5]. The altered pattern of gene expression leads to reprogramming of senCs metabolism and secretion of a wide range of soluble and insoluble factors which are commonly referred to secretory phenotype associated with aging (SASP, Senescence-Associated Secretory Phenotype) [6][7]. SASP includes many pro-inflammatory cytokines/chemokines, growth factors, metalloproteinases (MMPs), and other soluble factors (Figure 1) [8][9]. Due to secretion of these factors, cells with SASP by a “paracrine” way promote the aging of neighboring SCs, inflammation of the tissue, and destruction/degradation of dermal ECM through the action of MMPs. In addition to the soluble SASP factors, extracellular vesicles (exosomes) produced by senCs are also important mediators of the “paracrine” aging. By means of their constituent microRNAs, specific proteins, nucleic acids, and other insoluble factors, senCs perform intercellular connections, in particular between senDFs and neighboring SCs, enhancing the aging processes in these cells (as has been shown both in vitro and in vivo) [10].
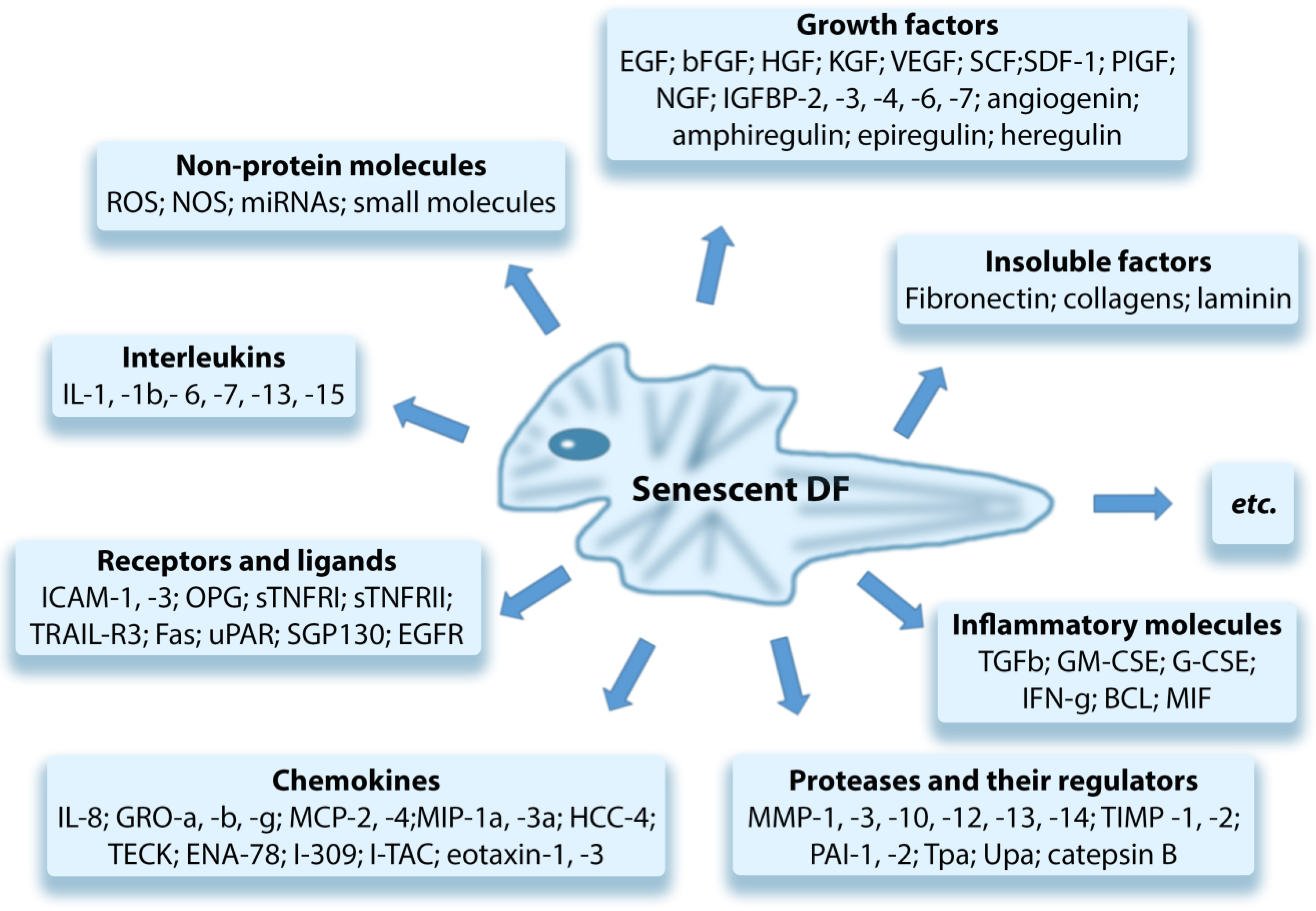
Figure 1. Components of the senescence-associated secretory phenotype (SASP).
Basisty et al. (2020) published the senDFs SASP atlas, which contains a large database, including the soluble factors and exosomes produced by senDFs. The authors revealed that SASP is variable and its composition may change depending on the trigger factor that causes cell aging [11]. Waldera Lupa et al. (2015), by examining the DFs obtained from the skin of elderly people with chronological aging, revealed a specific secretome, including 70 proteins corresponding to the classic SASP and 21 unique proteins that may reflect specific skin-aging processes [12].
2. Identification of Senescent Fibroblasts
Senescent DFs (like other senCs) are cells that do not have specific markers; so, they can be identified by means of a combination of characteristic features [13]. The following senDFs markers are most frequently used in vitro (they were initially studied in vitro during the induced DFs aging, both replicative and premature [14][15]):
- Morphological changes: the increase in size and flattening of the shape [16];
- Increased activity of senescence-associated lysosomal enzyme β-galactosidase (SA-b-gal) which is the “gold standard” for the identification of senCs both in vitro and in vivo (in tissue samples) [17];
- Increased levels of inhibitors of the cell cycle (p16INK4a, p21CIP1, and p53) [13][18];
- Visualization of cytoplasmic granularity under a light microscope: it indicates an increase in the number and size of lysosomes (while this does not mean an increase in the activity of these organelles since there is a marked decrease in the level of autophagy associated with lysosomes during aging) [19][20][21];
- Accumulation of lipofuscin [21];
- Increase in the frequency of γH2AX (a marker of double-stranded DNA breaks that occur during persistent DNA damage and DDR activation [13]);
- Presence of telomere-associated foci of DNA damage (TAF) [22];
- Decrease in the level of nuclear intermediate plate protein and epigenetic modulator of lamin B1 [23][24][25][26][27] (the level of lamin B1 decreases in vitro in senDFs regardless of the stress factor [25] and in vivo in DFs isolated from skin samples with signs of premature and chronological aging [28]);
- Presence of senescence-associated heterochromatin foci (SAHF, special heterochromatin structures formed in the nuclei of senCs) [29];
- Presence of DNA foci with chromatin changes that enhance cell aging (DNA-SCARS, DNA Segments with Chromatin Alterations Reinforcing Senescence) [30];
- Presence of HMGB1 (a protein from the group of nuclear non-histone proteins; in senCs it leaves the nucleus and moves to the cytoplasm and ECM; a decrease in its level in the nucleus leads to a decrease in gene expression) [31];
- High levels of proinflammatory cytokines in cells with SASP (in particular, IL-1α, IL-6, TNF, NF-κB, etc.), chemokines (CXCR2), metalloproteinases (MMP-3 and MMP-9), etc. [32][33][34];
- Mt dysfunction [35].
The DFs aging markers detected in vitro were also identified in vivo in the study of people with chronological aging [36][37] and photoaging [38]. Among these markers, the most common markers are the following:
- High level of SA-β-gal activity;
- Change in the production of ECM components;
- Increased level of cyclin-dependent kinases p21 and p16INK4a (among the other markers detected in vivo, it has the highest correlation with markers revealed in vitro [18]);
- Depletion of lamin B1 [25];
- Presence of SAHF (Senescent-Associated Heterochromatin Foci) [14][39];
- Increased level of SASP proinflammatory cytokines [33];
- Presence of telomere-associated foci of DNA damage (TAF) used as the quantitative marker of skin tissue aging in situ [22].
In addition, the high level of VEGF (vascular endothelial growth factor) can serve as one of the markers of senDFs in vivo, which is associated with increased vascular permeability, erythema, and the risk of skin cancer. It should be noted that the activation of VEGF production in senDFs does not depend on the type of cellular aging trigger [40]. In the scientific literature, much attention was paid to the identification of senDFs in the skin since there is no doubt about the key role of these cells in the induction and progression of skin-tissue aging [41][42][43][44][45]. According to Gorgoulis et al. (2019), for the most reliable identification of senCs in tissues (in situ), a “multi-marker approach” should be used, combining, for example, cytoplasmic markers (SA-β-gal and lipofuscin) with nuclear markers (p16INK4A and p21WAF1/Cip1), as well as the markers associated with SASP [8]. The most frequently used methods are the following: single cell analysis, enzyme immunoassay, visualizing flow cytometry, or mass cytometry [8].
3. The Role of senDFs in Skin Aging
It has been revealed that, with age, an accumulation of senDFs occurs in the skin, which, through the associated specific SASP and by paracrine mechanisms, contributes to depletion of the SC pool, destruction/disruption of the regenerative abilities of the tissue, and, therefore, the induction and progression of its aging [42][45][46][47][48][49][50][51].
It should be noted that senCs belong to a specific type of cells. The results of preclinical studies have shown that selective removal of senCs significantly improves tissue homeostasis, prolongs the life of animals, and improves life quality [3][52]. However, it has also been shown that removal of senCs from the wound delays the healing, leads to fibrosis, and impairs the formation of granulation tissue [53]. In addition, it has been established that senCs participate in the remodeling/plasticity of body tissues [3]. These effects are related to the fact that cellular aging is an evolutionary antagonistic pleiotropic process while senCs are involved in every stage of ontogenesis: from embryogenesis, where they participate in the development of organs/tissues, to a mature state, where they play a significant role in the repair of organs/tissues by preventing the proliferation of cells with damaged DNA, thereby lowering the risk of neoplastic transformation [2][3][54].
In young people, the immune system effectively eliminates senCs from the body. However, the functions of the immune system decrease with age and the accumulation of senescent cells is observed in tissues [55]. As a result, the destruction of tissues occurs due to a number of pathophysiological processes mediated by SASP not only at the level of tissues but also at the level of cell populations.
At the level of tissues, degradation of the ECM is observed, caused by overproduction of MMPs in senescent cells [56]. Mild chronic aseptic inflammation also develops due to the secretion of many pro-inflammatory factors by senescent cells [46], including cytokines (IL-1, IL-6, and IL-8) and tumor necrosis factor (TNF), which recruit macrophages, neutrophils, and T cells infiltrating the tissues [2][57][58]. Thus, for example, it has been shown that IL-1 is directly associated with “paracrine” aging in vivo through activation of the inflammasome complex (a multi-protein oligomeric complex in myeloid cells responsible for activation of the inflammatory response) [49]. As a result, the following events occur: (1) degradation of membrane receptors, signaling pathways, proteins, and other components of the ECM; (2) changes in functions of the SCs niches; (3) disruption of autophagy processes; and (4) activation of transcription factor NF-κB (transcription nuclear factor of activated B cells), promoting the progression of inflammation in tissues [59]. The tissue inflammation is also accompanied by high levels of proinflammatory cytokines (IL-6, IL-1β, TGF-b, and TNF-α) that disrupt the transmission of anabolic signals, which leads to a decrease in the sensitivity of tissues to nutrients [60]. This close connection between aging and the inflammatory processes has been called “inflammaging” (inflammation + aging) or inflammatory aging.
At the level of cell populations, there are changes that are primarily associated with the paracrine mechanism, causing the aging phenotype in SCs located in spatial proximity to senCs [48][51][60][61][62][63][64], the so-called passive senescence [61] or “aging of an outsider observer” [62]. At the same time, the increase in the level of ROS is observed in senDFs, which causes the increase in Mt dysfunction. Thus, a vicious circle is formed (for more information, see above) that contributes to an even greater increase in ROS production and promotes the damaging effect of ROS on cell structures/organelles, which is accompanied by the reduced activity of antioxidant enzymes that is observed in cells with age [64][65]. In addition, factor NF-κB is activated and the “axis” ROS–NF-κB is formed, inducing DDR in SCs adjacent to senCs (the so-called “witness cells”) [42][63][66][67], which leads to the subsequent cell apoptosis or aging. At the same time, AP-1 and NF-B-dependent signaling pathways are also activated in senDFs, which contribute to the progression of inflammation in tissues [68].
Summarizing the abovementioned data and taking into account the main negative effects of senCs occurring in the skin [42][63], one can conclude the following about senescent cells:
- They do not proliferate, which leads to violation of the SCs’ self-renewing process and depletion of the SCs pool;
- Cause aging of neighboring SCs;
- Promote an increase in the level of ROS and cause Mt dysfunction;
- Induce DNA damage and aging of “witness cells” through the paracrine mechanism and ROS overproduction;
- Cause the chronic aseptic inflammation in tissues due to the effect of proinflammatory SASP factors secreted by senCs;
- Enhance the ECM degradation in the dermis by producing the high MMPs level;
- Disrupt cellular and tissue homeostasis.
Thus, the pathogenetic processes associated with the accumulation of dysfunctional senDFs in skin tissues with age lead to depletion of the SC pool and, therefore, to the decrease in regenerative abilities and the progression of aging processes in the tissues (Figure 2) [42][50][62][64][66]. The main research data confirming the role of senDFs in skin aging are the following [42][43][69]:
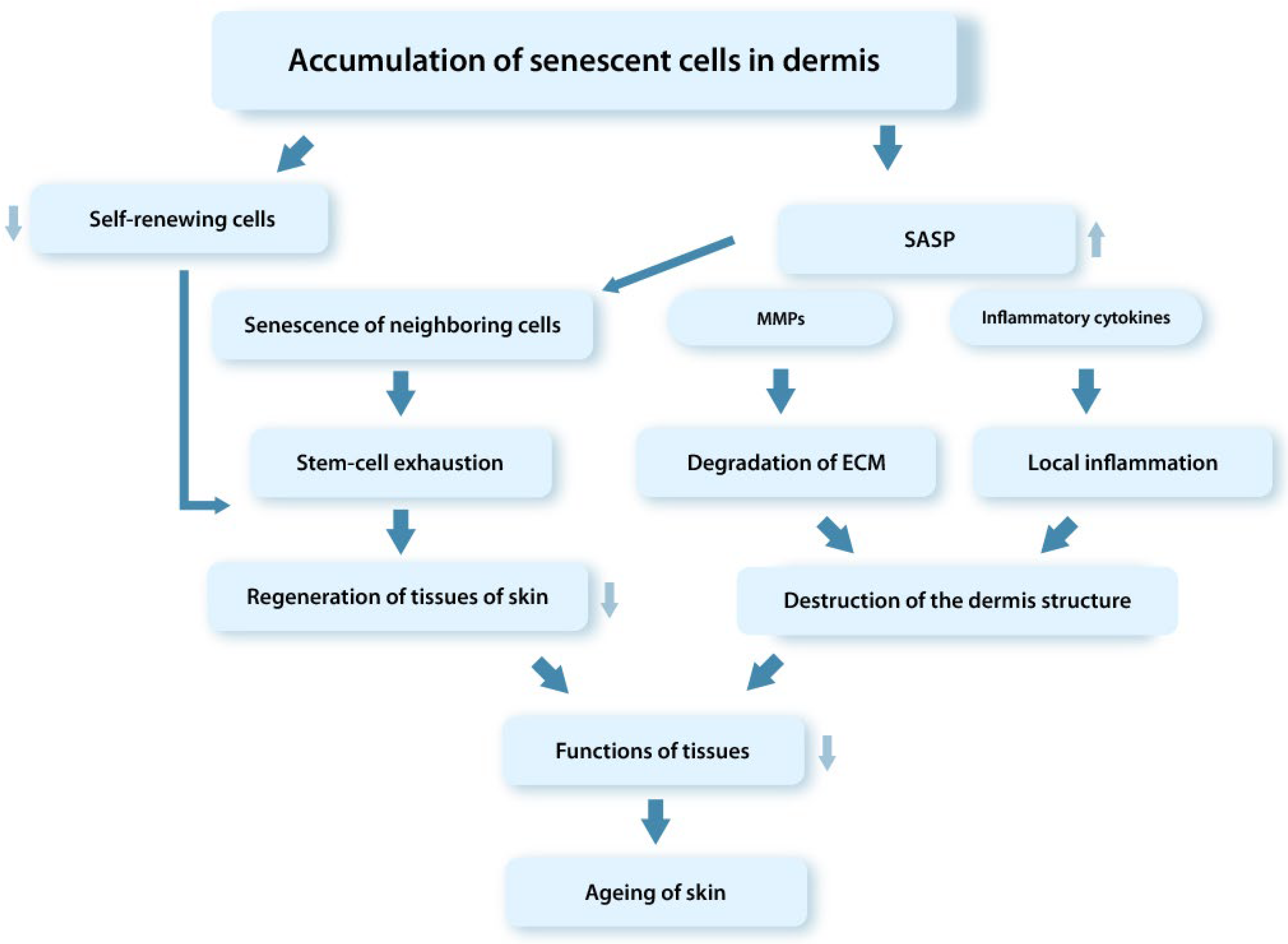
Figure 2. Schematic representation of mechanisms of the tissue dysfunction involving senCs (SASP). SenCs accumulating with age in tissues contribute through the SASP factors to the depletion of the SC pool, degradation of ECM in the dermis, development of the chronic aseptic inflammation in tissues, and disruption of the tissue regenerative potential.
- An increase in the level of p16INK4a-positive DFs in the dermis correlating with the formation of wrinkles and the appearance of typical signs of elastic fiber aging;
- During chronological aging, DFs have a proteomic profile in situ identical to the senDFs profile;
- An increase in the ROS level leads to an increase in the number of p16INK4a-positive DFs in the skin and correlates with the progression of skin aging;
- The spread of senescence to neighboring DFs with the expression of characteristic markers of cellular aging was recorded during transplantation of human senDFs into the skin of young immunodeficient mice;
- Organ cultures obtained on the basis of human senDFs have signs of aging typical for chronological aging of the skin, including impairment of epidermal morphogenesis;
- DFs isolated from the skin of elderly people are characterized by a gene expression pattern similar to that of senDFs;
- Studies using a model of perforin-deficient mice (characterized by reduced functions of NK cells) have demonstrated the suppressed ability of the immune system to eliminate senDFs which by accumulating in the dermis lead to structural changes and the progression of aging processes in the dermis;
- The results of a clinical study conducted using the local application of rapamycin on the skin of elderly people (with chronological aging) showed a decrease in the level of p16INK4a-positive DFs, as well as a decrease in the number of fine wrinkles and an increase in the thickness and elasticity of the skin.
- Rapamycin is an inhibitor of mTOR (protein regulating the cell cycle and participating in the aging of DFs through the regulation of SASP) suppressing the translation of membrane-bound cytokine IL-1a and thereby inhibiting the secretion of pro-inflammatory SASP factors induced by IL-1a.
References
- Calcinotto, A.; Kohli, J. Cellular senescence: Aging, cancer, and injury. Physiol. Rev. 2019, 99, 1047–1078.
- Munoz-Espin, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496.
- Yosef, R.; Pilpel, N. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 2016, 7, 11190.
- Baar, M.P.; Brandt, R.M. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 2017, 169, 132–147.
- Pereira, B.I.; Devine, O.P. Senescent cells evade immune clearance via HLA-E-mediated NK and CD8(+) T cell inhibition. Nat. Commun. 2019, 10, 2387.
- Demaria, M.; Desprez, P. Cell Autonomous and Non-autonomous Effects of Senescent Cells in the Skin. J. Investig. Dermatol. 2015, 35, 1722–1726.
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246.
- Gorgoulis, V.; Adams, P.D. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 814–827.
- Rodier, F.; Coppe, J.-P. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979.
- Borghesan, M.; Fafián-Labora, J. Small Extracellular Vesicles Are Key Regulators of Non-cell Autonomous Intercellular Communication in Senescence via the Interferon Protein IFITM. Cell Rep. 2019, 27, 3956–3971.
- Basisty, N.; Kale, A. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020, 18, e3000599.
- Waldera Lupa, D.M.; Kalfalah, F. Characterization of skin aging-associated secreted proteins (SAASP) produced by dermal fibroblasts isolated from intrinsically aged human skin. J. Investig. Dermatol. 2015, 135, 1954–1956.
- Wang, A.S.; Dreesen, O. Biomarkers of cellular senescence and skin aging. Front. Genet. 2018, 9, 247.
- Waaijer, M.; Parish, W.E. The number of p16INK4a positive cells in human skin reflects biological age. Aging Cell 2012, 11, 722–725.
- Gruber, F.; Kremslehner, C. Cell aging and cellular senescence in skin aging—Recent advances in fibroblast and keratinocyte biology. Exp. Gerontol. 2020, 130, 110780.
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965, 37, 614–636.
- Dimri, G.P.; Lee, X. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367.
- Sharpless, N.E.; DePinho, R.A. How stem cells age and why this makes us grow old. Nat. Rev. Mol. Cell Biol. 2007, 8, 703–713.
- Park, J.T. Adjustment of the lysosomal-mitochondrial axis for control of cellular senescence. Ageing Res. Rev. 2018, 47, 176–182.
- Ott, C.; König, J. Macroautophagy is impaired in old murine brain tissue as well as in senescent human fibroblasts. Redox Biol. 2016, 10, 266–273.
- Eckhart, L.; Tschachler, E. Autophagic control of skin aging. Front. Cell Dev. Biol. 2019, 7, 143.
- Hewitt, G.; Jurk, D. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 2012, 3, 708.
- Waaijer, M.E.C.; Gunn, D.A. Do senescence markers correlate in vitro and in situ within individual human donors? Aging 2018, 10, 278–289.
- Scaffidi, P.; Misteli, T. Lamin A-dependent nuclear defects in human aging. Science 2006, 312, 1059–1163.
- Freund, A.; Laberge, R.-M. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 2012, 23, 2066–2075.
- Taimen, P.; Pflegar, K. A progeria mutation reveals functions for lamin A in nuclear assembly, architecture, and chromosome organization. Proc. Natl. Acad. Sci. USA 2009, 106, 20788–20793.
- Dreesen, O.; Chojnowski, A. Lamin B1 fluctuations have differential effects on cellular proliferation and senescence. J. Cell Biol. 2013, 200, 605–617.
- Bernardes de Jesus, B.; Blasco, M.A. Assessing Cell and Organ Senescence Biomarkers. Circ. Res. 2012, 111, 97–109.
- Sun, L.; Yu, R. Chromatin architectural changes during cellular senescence and aging. Genes 2018, 9, 211.
- Narita, M.; Nũnez, S. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716.
- Davalos, A.R.; Kawahara, M. p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J. Cell Biol. 2013, 201, 613–629.
- Tilstra, J.S.; Robinson, A.R. NF-κB inhibition delays DNA damage-induced senescence and aging in mice. J. Clin. Investig. 2012, 122, 2601–2612.
- Coppe, J.P.; Desprez, P.Y. The senescence-associated secretory phenotype: The dark side of tumor suppression. Ann. Rev. Pathol. 2010, 5, 99–118.
- Acosta, J.C. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008, 133, 1006–1018.
- Korolchuk, V.I.; Miwa, S. Mitochondria in cell senescence: Is mitophagy the weakest link? EBioMedicine 2017, 21, 7–13.
- Ressler, S.; Bartkova, J. p16INK4A is a robust in vivo biomarker of cellular aging in human skin. Aging Cell 2006, 5, 379–389.
- Campisi, J. Aging, Cellular Senescence, and Cancer. Ann. Rev. Physiol. 2013, 75, 685–705.
- Debacq-Chainiaux, F.; Leduc, C. UV, stress and aging. Dermato-Endocrinol. 2012, 4, 236–240.
- Aird, K.M.; Zhang, R. Detection of senescence-associated heterochromatin foci (SAHF). Methods Mol. Biol. 2013, 965, 185–196.
- Coppe, J.P.; Kauser, K. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J. Biol. Chem. 2006, 281, 29568e74.
- Jenkins, G. Molecular mechanisms of skin ageing. Mechanisms of Ageing and Development. Mech. Ageing Dev. 2002, 123, 801–810.
- Wlaschek, M.; Maity, P. Connective tissue and fibroblast senescence in skin aging. J. Investig. Dermatol. 2021, 141, 985–992.
- Fitsiou, E.; Pulido, T. Cellular senescence and the senescence-associated secretory phenotype as drivers of skin photoaging. J. Investig. Dermatol. 2020, 141, 1119–1126.
- Velarde, M.C.; Demaria, M. Targeting senescent cells: Possible implications for delaying skin aging: A mini-review. Gerontology 2016, 62, 513–518.
- Ho, C.-Y.; Dreesen, O. Faces of cellular senescence in skin aging. Mech. Age Dev. 2021, 198, 111525.
- Cinat, D.; Coppes, R.P. DNA Damage-Induced Inflammatory Microenvironment and Adult Stem Cell Response. Front. Cell. Dev. Biol. 2021, 9, 729136.
- Childs, B.G.; Durik, M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435.
- Da Silva, P.F.L.; Schumacher, B. DNA damage responses in ageing. Open Biol. 2019, 9, 190168.
- Acosta, J.C.; Banito, A. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990.
- Tchkonia, T.; Zhu, Y. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Investig. 2013, 123, 966–972.
- Toutfaire, M.; Bauwens, E. The impact of cellular senescence in skin ageing: A notion of mosaic and therapeutic strategies. Biochem. Pharmacol. 2017, 142, 1–12.
- Baker, D.J.; Childs, B.G. Naturally occurring p16 Ink4a-positive cells shorten healthy lifespan. Nature 2016, 530, 184e9.
- Demaria, M.; Ohtani, N. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 2014, 31, 722.
- Muñoz-Espín, D.; Cañamero, M. Programmed cell senescence during mammalian embryonic development. Cell 2013, 155, 1104–1118.
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. Cell. Biol. 2018, 217, 65–77.
- Wiggins, K.A.; Clarke, M.C. Senescence utilises inflammatory caspases to drive the SASP. Aging 2019, 11, 3891–3892.
- Fafián-Labora, J.A.; O’Loghlen, A. NF-κB/ IKK activation by small extracellular vesicles within the SASP. Aging Cell 2021, 20, e13426.
- Kale, A.; Sharma, A. Role of immune cells in the removal of deleterious senescent cells. Immun. Ageing 2020, 17, 16.
- Salminen, A.; Kaarniranta, K. Inflammaging: Disturbed interplay between autophagy and inflammasomes. Aging 2012, 4, 166–175.
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446.
- Nelson, G.; Wordsworth, J. A senescent cell bystander effect: Senescence-induced senescence. Aging Cell 2012, 11, 345–349.
- Da Silva, P.F.L.; Ogrodnik, M. The bystander effect contributes to the accumulation of senescent cells in vivo. Aging Cell 2019, 18, e12848.
- Nelson, G.; Kucheryavenko, O. The senescent bystander effect is caused by ROS-activated NF-kB signaling. Mech. Ageing Dev. 2018, 170, 30–36.
- Faget, D.V.; Ren, Q. Unmasking senescence: Context-dependent effects of SASP in cancer. Nat. Rev. Cancer 2019, 19, 439–453.
- Tomaru, U.; Takahashi, S. Decreased proteasomal activity causes age-related phenotypes and promotes the development of metabolic abnormalities. Am. J. Pathol. 2012, 180, 963–972.
- Lee, Y.; Choi, S. Cellular Senescence and Inflammaging in the Skin Microenvironment. Int. J. Mol. Sci. 2021, 22, 3849.
- Krutmann, J.; Schroeder, P. Role of mitochondria in photoaging of human skin: The defective powerhouse model. J. Investig. Dermatol. Symp. Proc. 2009, 14, 44–49.
- Meyer, P.; Maity, P. A model of the onset of the senescence associated secretory phenotype after DNA damage induced senescence. PLoS Comput. Biol. 2017, 13, e1005741.
- Laberge, R.M.; Sun, Y. mTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting il1a translation. Nat. Cell Biol. 2015, 17, 1049–1061.
More
Information
Subjects:
Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.6K
Revisions:
2 times
(View History)
Update Date:
16 Jun 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No