 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Italia Falcone | + 5252 word(s) | 5252 | 2020-10-07 04:29:50 | | | |
| 2 | Bruce Ren | -13 word(s) | 5239 | 2020-10-10 03:45:25 | | | | |
| 3 | Bruce Ren | Meta information modification | 5239 | 2020-10-21 09:49:33 | | |
Video Upload Options
Antitumor therapies have made great strides in recent decades. Chemotherapy, aggressive and unable to discriminate cancer from healthy cells, has given way to personalized treatments that, recognizing and blocking specific molecular targets, have paved the way for targeted and effective therapies. Melanoma was one of the first tumor types to benefit from this new care frontier by introducing specific inhibitors for v-Raf murine sarcoma viral oncogene homolog B (BRAF), mitogen-activated protein kinase (MEK), v-kit Hardy–Zuckerman 4 feline sarcoma viral oncogene homolog (KIT), and, recently, immunotherapy. However, despite the progress made in the melanoma treatment, primary and/or acquired drug resistance remains an unresolved problem. The molecular dynamics that promote this phenomenon are very complex but several studies have shown that the tumor microenvironment (TME) plays, certainly, a key role. In this review, we will describe the new melanoma treatment approaches and we will analyze the mechanisms by which TME promotes resistance to targeted therapy and immunotherapy.
1. Introduction
Melanoma is one of most aggressive human tumors, arising from the uncontrolled proliferation of melanocytes, the skin cells responsible for the production of melanin. In terms of incidence, malignant melanoma accounts for approximately 5% of all malignant tumors and its incidence is highly variable, depending on race and geographical variations: It is predominantly diagnosed in Caucasians and 85% of cases occur in North America, Europe, and Oceania. Although highly curable when diagnosed in an early phase, melanoma is an aggressive disease with five years’ relative survival of only 25%, when diagnosed at an advanced metastatic stage [1][2]. Like many other solid tumors, malignant melanoma is highly heterogeneous and substantially resistant to unselective treatments, such as chemotherapy. In the past few years, mutational analysis and next-generation sequencing (NGS) approaches have shown that somatic mutations in BRAF or neuroblastoma RAS viral oncogene homolog (NRAS) genes promote deregulated survival and migration when combined with genetic alterations and/or epigenetic events that support senescence bypass [3][4].
Moreover, metastatic melanoma is considered a perfect example of immunogenic tumor because it is characterized by the consistent presence of lymphocid infiltrate, as compared to other cancers [5]. Based on these observations, molecularly targeted therapy and immunotherapy have revolutionized the approach to melanoma treatment and overall management. Clinical evidence has shown extremely encouraging results in terms of overall survival (OS) in patients treated with targeted therapy and immunotherapy [6][7][8][9]. Despite many important advances, however, development of the resistance remains a significant obstacle to melanoma curability and can be modulated by several factors, both intrinsic and extrinsic to the cancer cell. One such important factor is certainly the tumor microenvironment (TME), an intricate and complex network of cells, molecules, and paracrine factors that are tightly interconnected with melanoma cells, thereby influencing their initiation, progression, and sensitivity/resistance to therapeutic interventions.
2. TME Implications in Drug Resistance for Melanoma
Development of therapeutic resistance arguably represents the most important challenge in cancer therapy. Such phenomenon is associated with disease progression and low survival rates and is promoted by the ability of cancer cells to activate both intrinsic (i.e., dependent on genetic changes occurring in the cancer cell itself) and extrinsic (i.e., mediated by cross-talk mechanisms occurring between cancerous and noncancerous cells) escape mechanisms. Tumors are characterized by high genomic instability and heterogeneity and these prerogatives may lead to both primary (or de novo) or acquired resistance (i.e., occurring in cells previously responsive to the same treatment). Most importantly, the selective pressure applied by treatment itself may select out specific mechanisms of resistance. In some instances, therapeutic resistance occurs independent of genetic changes modifying cancer cells’ acquired capabilities: In these cases, the insurgence of drug resistance can be attributed to changes occurring in different compartments of the TME. Indeed, tumor masses, including those that form in metastatic melanoma, should not be considered as isolated contexts without interactions; indeed, it has long been known that tumor cells "cross talk" continuously with many cellular and acellular components of the tumor stroma, which surrounds and penetrates the tumor mass. Such intricate structures constitute the TME and are characterized by mutual and continuous interactions between tumor and nontumor cells. TME is composed of cells and extracellular components of different origins, which contribute in several ways to the various stages of tumor progression (Figure 2) [10][11].
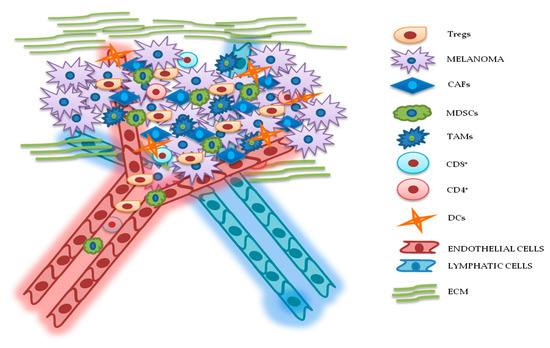
Figure 2. Relationship between melanoma and tumor microenvironment (TME). In this figure, is illustrated schematically the reciprocal interactions between melanoma cells and the other components of TME. Melanoma’s TME, involved in tumor growth, progression, and drug resistance, is essentially represented by regulatory T cells (Tregs), cancer-associated fibroblasts (CAFs), myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), cluster differentiation 4 (CD4+)/CD8+ lymphocytes, dendritic cells (DCs), endothelial and lymphatic cells, and extracellular matrix (ECM).
3.1. Cellular Components
3.1.1. Cancer-Associated Fibroblasts (CAFs)
Fibroblasts are important components of the stroma and their physiological functions encompass synthesis of extracellular matrix (ECM) and regulation of the inflammatory process. Upon tight (direct or mediated by soluble factors) interaction with cancer cells, fibroblasts differentiate into CAFs, which are characterized by specific markers: α smooth muscle actin (α-SMA), fibroblast activation protein (FAP), vimentin, fibroblast specific protein 1 (FSP1), and platelet-derived growth factor receptor (PDGFR)-α and β [12][13][14]. CAFs are involved in many cellular processes, including ECM remodeling, angiogenesis, and cell-to-cell interactions; in vivo, their activation is fundamental for tumor neo-vascularization [15][16]. In vitro and in vivo studies have shown that the continuous and persistent interactions between tumor cells and CAFs promote many aspects of the tumorigenic process, such as tumor progression, metastasis, and drug resistance. Melanoma cells, co-cultured with CAFs or grown in their conditioned media, display greater invasion and migration capabilities, as compared to the same cells cultured in isolation [17][18]. Recent studies also confirm that CAFs’ activation is probably a crucial step for melanoma metastasis formation. Indeed, mice, in which the CAFs are inhibited by β-catenin suppression, displayed markedly decreased tumor-mediated vascularization [19]. CAFs and tumor cells reciprocally influence each other’s biological behavior, and such cross talk is finely regulated by specific molecular mechanisms. As described in Figure 3A, the co-regulation system can involve tumor necrosis factor receptor-associated factor 6 (TRAF6), expressed in CAFs’ activated and melanoma cells [20].
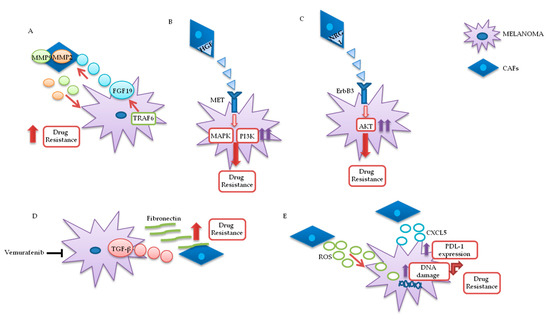
Figure 3. Melanoma/CAFs’ paracrine interconnections. (A–E) In this figure, is illustrated schematically the mutual interactions between melanoma cells and cancer-associated fibroblasts (CAFs). Several factors are implicated in these intricate interconnections at the basis of drugs resistance: Tumor necrosis factor receptor-associated factor 6 (TRAF6), fibroblast growth factor 19 (FGF19), metalloproteinases 2 and 9 (MMP2 and MMP9), hepatocyte growth factor (HGF), neuregulin 1 (NRG1), V-erb-b2 avian erythroblastic leukemia viral oncogene homolog3 (ErbB3), transforming growth factor β (TGF-β), reactive oxygen species (ROS), CXC motif chemokine 5 (CXCL5), programmed death-ligand 1 (PDL-1).
In melanoma cells, TRAF6 promotes nuclear factor kappa-light-chain-enhancer of activated B cells (NFkB)-dependent release of fibroblast growth factor 19 (FGF19), implicated in the transformation and activation of fibroblasts. FGF19-mediated CAFs’ activation supports, in turn, the malignant and invasive phenotype of melanoma cells and their drugs resistance. On the other hand, TRAF6 upregulation in fibroblasts results in ECM remodeling through the release of matrix metalloproteinases (MMPs) 2 and 9 .
The mutual interaction between melanoma and CAFs can promote drug resistance in different ways. Straussman and collaborators highlighted the role of hepatocyte growth factor (HGF) in the development of acquired resistance to BRAF inhibitors (Figure 3B). Co-culture systems and proteomics analysis showed that HGF secreted by fibroblasts, by interacting with its mesenchymal ephitelial transition (MET) receptor on melanoma, induced MAPK and PI3K pathways’ activation, thereby promoting resistance to RAF inhibition. Simultaneous downregulation of both RAF and MET reverted resistance in vitro, and it has been proposed as a possible therapeutic approach for the treatment of BRAF-mutant melanomas. Most importantly, the authors confirmed increased HGF expression in stromal cells of BRAF-mutant melanoma patients undergoing BRAF-targeted treatment in vivo, which resulted in poor prognosis and decreased response to treatments [21]. Neuregulin 1 (NRG1) is another paracrine factor through which CAFs may influence melanoma response to MAPK inhibitors (Figure 3C). NRG1 is the ligand of v-erb-b2 avian erythroblastic leukemia viral oncogene homolog3 (ErbB3), which is upregulated in melanoma cells after treatment with BRAF inhibitors. The use of ErbB3/ErbB2 antibodies restores the cytotoxic activity of these drugs in BRAF-mutant melanoma cell lines [22]. Furthermore, vemurafenib treatment increases the production of transforming growth factor β (TGF-β) by melanoma cells; TGF-β, in turn, causes CAFs’ activation and increased fibronectin production, involved in BRAF inhibitors’ resistance (Figure 3D) [23].
Moreover, paradoxical MAPK activation, induced by BRAF inhibitors in genetically “normal” stromal cells, promotes a “therapy-resistant” microenvironment: Intravital imaging analyses conducted in melanoma have shown major paradox MAPK reactivation, especially in areas with high stromal density. Such activated CAFs, in turn, promoted matrix remodeling and ERK reactivation in melanoma, through integrin β1/focal adhesion kinase (FAK)/v-src sarcoma (Schmidt–Ruppin A-2) viral oncogene homolog avian (Src) signaling [24].
Uncontrolled production of reactive oxygen species (ROS) by TME fibroblasts is also associated with resistance to BRAF-targeted agents in melanoma (Figure 3E). Aging fibroblasts tend to release high levels of ROS in the TME, thereby modulating MAPK and PI3K pathways’ activation in tumor cells and promoting cells’ growth and drug resistance [25]. Such phenomenon could potentially be reversed by treating melanoma cells with antioxidants, thereby restoring drug responsiveness[26]. Along these lines, a recent preclinical study analyzed the role of secreted frizzled-related protein 2 (sFRP2), a wingless type MMTV integration site family member (Wnt) antagonist, in vemurafenib resistance: The sFRP2, produced and released in the TME by aged fibroblasts, actives a cascade of events in melanoma cells, ultimately leading to the loss of the redox effector apurinic/apyrimidinic endonuclease 1 (APE1). This condition significantly reduces the ability of melanoma cells to overcome ROS-induced DNA damage. Moreover, sFRP2-mediated inhibition of β-catenin leads to reduced melanoma response to vemurafenib .
It has recently been shown that CAFs are involved in the induction of a protumor immune microenvironment in many cancer models, favoring tumor growth and pharmacological resistance [27][28][29]. CAFs-mediated CXC chemokine ligand CXCL-2 production promotes regulatory T cells’ (Tregs) growth and recruitment in the tumor stroma [29[30]. Melanoma-associated fibroblasts also directly influence tumor cells’ ability to adapt and modify the response to immunotherapy. Experiments conducted on melanoma cell cultures have shown that CAFs release CXC motif chemokine 5 (CXCL5), which, in turn, induces PI3K/ protein kinase B (AKT)-dependent PDL-1 expression and resistance to immunotherapy in melanoma cells (Figure 3E) [31]. In addition, TGF-β secreted by CAFs also promotes resistance to PD-1 inhibitors: Transcriptomic and flow cytometric analysis, conducted on biopsies from 94 melanoma patients at various treatment stages, revealed a subset of patients characterized by loss of major histocompatibility complex 1 (MCH-I) and disease progression: Such phenomenon, induced by TGF-β released by CAFs, promotes a microphthalmia-associated transcription factor (MITF)low/AXLhigh phenotype in melanoma cells, associated with resistance to MAPK pathway and PD-1 inhibitors [32].
Finally, CAFs’ involvement in drug resistance is not limited to the production of paracrine factors but is also associated with cell-to-cell contact with cancer cells. Several studies have shown that fibroblasts create a physical barrier around the tumor mass and directly activate survival pathways in cancer cells [33][34][35]. In particular, these interactions are promoted by N-cadherin, expressed by both melanoma cells and fibroblasts, and are involved in tumor activation of the survival pathway PI3K/AKT/ BCL2 associated agonist of cell death (BAD) [33].
3.1.2. Lymphocytes
The immuno-microenvironment is characterized by T lymphocytes that recognize antigenic peptides presented by other components of the immune system [36]. CD4+ T cells act as immune response “adjuvants” through the secretion of specific cytokines. CD8+ T lymphocytes, on the other hand, are responsible for direct antigen/tumor cell individuation/elimination and are considered the most important mediators of tumor immune surveillance [37].
Depending on their genetic background, melanoma cells can influence the development of an immunosuppressive microenvironment. Phosphatase and tensin homolog deleted on chromosome 10 (PTEN), for example, is an important tumor suppressor gene often mutated/deleted in several cancer types, including melanoma; indeed, PTEN loss is present and concomitant with BRAF mutations in about 44% of melanomas and is associated with reduced OS [38]. PTEN loss promotes the formation of TME with low levels of cytotoxic T and natural killer (NK) cells and high concentrations of immunosuppressive elements, such as myeloid-derived suppressor (MDSCs) Tregs cells [39]. As described in in vitro and in vivo studies, PTEN-null melanoma cells inhibit antitumor activity of T cells and, consequently, response to immunotherapy. Through its negative regulation of PI3K and signal transducer and activator of transcription (STAT) 3 pathways, PTEN inhibits the production of immunosuppressive cytokines, such as interleukins (IL) 6 and 10 and vascular endothelial growth factor (VEGF). In melanoma, PTEN loss promotes STAT3 activation and, consequently, overproduction of these cytokines [40]. Moreover, PTEN loss is associated with reduced T cells’ recruitment to the tumor site and cytotoxic activity [41].
An immunosuppressive TME influences the differentiation of dysfunctional CD8+ T lymphocytes, i.e., T cells with reduced growth and effectors’ cell recognition capacity and high concentrations of PD-1 and CTLA-4 receptors. If physiological conditions such as this status are necessary for immune homeostasis and to avoid self-reactive phenomena, in tumor contexts it may be an escape route that cancer cells use to evade immune response and promote resistance to immunotherapy [42]. A study conducted in patients with advanced melanoma demonstrated the presence of a subpopulation of T cells with high levels of PD-1 and immunoglobulin and mucin domain-containing molecule 3 (Tim3), another inhibitory receptor. Tim3 inhibition partially reverted the dysfunctional condition of T cells and increased their antitumor abilities. These results form the rationale for simultaneous blockade of PD-1 and Tim3 as a possible therapeutic approach to restore CD8+ T lymphocytes’ functionality in context of melanoma [43]. The same research group identified an additional inhibitory receptor, called T cell immunoglobulin (Ig) and immunoreceptor tyrosine-based inhibition motif (ITIM) domain (TIGIT). Inhibition of this receptor together with PD-1 may counteract dysregulated T cells’ activity in a manner similar to Tim3 inhibition [44]. Extensive transcriptional profiling of the tumor infiltrate in 25 melanoma patients recently showed clonal expansion of dysfunctional CD8+ T cell subset. The authors highlighted the reactivity and differentiation of these cells, which are likely involved in the regulation of antitumor activity and resistance to immunotherapeutic agents, making them an attractive target for more targeted and effective immunotherapeutic treatments in melanoma [45].
B lymphocytes are the cells responsible for humoral and acquired immunity. Their main function is to produce specific antibodies against foreign antigens, but they are also involved in maintenance of immune memory [46]. In melanoma, tumor-associated B cells (TAB) account for up to 33% of TME immune cells and are involved in resistance to targeted therapy by promoting angiogenesis and chronic inflammation. In addition, the presence of B cells in the tumor infiltrate is associated with increased metastatic capacity of melanoma cells and reduced patients’ OS [47]. Recently, an interesting study analyzed the cross talk between melanoma cells and TAB and identified specific stimulating factors involved in the modulation of tumor response to different drugs. Melanoma secretes fibroblast growth factor 2 (FGF2), which actives B cells through its binding to fibroblast growth factor receptor 3 (FGFR-3) and promotes the release of insulin-like growth factor 1 (IGF-1). This factor, on tumor cells, induces proliferation and drug resistance. IGF-1, in turn, induces tumor cell proliferation and drug resistance. High levels of IGF-1 and FGFR-3 have been found in biopsies of melanoma patients treated with BRAF inhibitors in monotherapy or in combination with MEK inhibitors and IGF-1, and its receptor (IGF-1R) are associated with resistance to MAPK inhibitors [47]. However, TABs may have an opposite function in response to immunotherapy in melanoma. Indeed, a particular subtype of TABs can instead promote melanoma response to ICIs, by promoting the recruitment of CD8+ T cells in the tumor compartment. The authors observed that the presence of higher concentrations of these B cells, in pretreated melanoma patients, is associated with a better response to future immunotherapy treatments [48]. More recently, analysis of metastatic melanoma samples showed that the co-occurrence of tumor- associated CD8+ T cells and CD20+ B cells is associated with improved survival [49]. The formation of tertiary lymphoid structures in these CD8+/CD20+ tumors is associated with a gene signature, which predicts clinical outcomes in melanoma patients treated with ICIs. Moreover, B cell-rich melanomas displayed increased levels of transcription factor 7 (TCF7)+ naive and/or memory T cells, whereas T cells in tumors without tertiary lymphoid structures had a dysfunctional molecular phenotype. In another study, it was shown that B cell signatures are enriched in human melanoma samples from patients who responded to neoadjuvant ICI treatment [50]. B cell markers were, indeed, the most differentially expressed genes in the tumors of responders versus non responders [51]. Histological evaluation again highlighted the localization of B cells within tertiary lymphoid structures, while RNA sequencing demonstrated clonal expansion and unique functional states of B cells (switched memory B cells) in responder.
NKs are an important subclass of granular lymphocytes, involved in the recognition and elimination of virus-infected and transformed cells [52][53]. In general, cancer promotes several mechanisms that destabilize the functionality of NKs, determining immune evasion: (1) Hyperproduction of activating ligands that paradoxically block NKs’ receptors and (2) release of immunosuppressive factors, such as TGF-β and prostaglandin E [54]. Moreover, vemurafenib treatment of melanoma cells induces suppression of NKs activity in vitro, through downregulation of natural killer group 2D (NKG2D) and DNAX accessory molecule-1 (DNAM-1) activating receptors and simultaneous upregulation of major histocompatibility complex (MHC) I, which plays an inhibitory effect on NK cells [55].
Tregs represent a CD4+ T cell’s subpopulation with immunosuppressive properties [56][57]. In different cancer types, including melanoma, Tregs are able to promote immune evasion and cancer progression and are associated with poor prognosis [58][59][60]. In an analysis conducted on peripheral blood mononuclear cells (PBMCs) collected from healthy volunteers, Baumgartner and collaborators observed that melanoma evades the immune system by activation of Treg cells. Indeed, PBMCs exposed to melanoma-conditioned medium for a week presented an increase in Tregs’ induction and a major presence of IL-10 and TGF-β in the supernatant, as compared to the same PBMCs grown in control medium [61]. In BRAF-mutant melanomas, uncontrolled MAPK activation leads to an increased production of different ILsand VEGF that influence the activity of the immune system toward a protumor condition. Sumimoto and collaborators showed that in BRAF-mutant melanomas Tregs are activated and suppress the antitumor function of T lymphocytes. Moreover, pharmacological blockades or genetic manipulation of key components of MAPK pathway drastically decrease tumor production of immunosuppressive cytokines, allowing for the development of an immune microenvironment favorable to tumor suppression [62].
Regulation of Tregs’ differentiation and function could, therefore, be considered a valid therapeutic target for many cancers, including melanoma. Tregs are characterized by constitutive upregulation of PD-1 and CTLA-4 receptors and this condition leads to the hypothesis that Tregs could be the actual targets of ICI-based immunotherapy [63]. Unfortunately, results obtained in different studies are conflicting. Indeed, some studies have confirmed the inhibitory action of ICI on Tregs’ functionality, while others have reported opposite results that could support the hypothesis of an involvement of ICI-mediated activation of these cells in immune-resistance [64][65][66]. Analysis conducted on murine models of autoimmune pancreatitis have partly elucidated the suppressive role of PD-1 on Treg cells activity. Indeed, mice characterized by PD1-deficient Tregs showed greater immunosuppressive capacities and rapid development of autoimmune disease [64]. On the basis of these results, it can be speculated that, physiologically, the PD-1 axis plays an important role in the regulation of Tregs’ functionality and its inhibition may result in their increased activity. In vitro and in vivo experiments showed that, after treatments with nivolumab, Tregs proliferate and are functionally activated, resulting in the inhibition of antitumor activity [66]. Although with somewhat conflicting results, anti-CTLA-4 therapy would seem to bring more favorable effects on Tregs inhibition. Melanoma patients treated with ipilimumab showed a reduction in Tregs’ levels and major benefits in terms of decreased tumor growth and survival [67]. In mice models of melanoma, CTLA-4 blockade increases the intratumor effector T cells/Tregs ratio, through fragment crystallizable (Fc)-gamma receptor (FcγR)-dependent mechanism. FcγR is expressed by several immune cells, such as macrophages, neutrophils and NK cells and, therefore, TME composition may influence the response to CTLA-4 inhibitors. Melanomas presenting low concentrations of macrophages or immune cells deficient for FcγR tend to respond less to therapy [68].
3.1.3. MDSCs
MDSCs are immature myeloid cells, with immunosuppressive functions, associated to tumor progression, metastasis, angiogenesis, and drug resistance. Absent or present in small concentrations in physiological conditions, they are recruited by tumor cells or by inflammatory stimuli and are responsible for the production of several factors involved in tumor growth and immune evasion, such as IL-10, TGF-β, and VEGF [69][70]. In melanoma, chronic inflammation promotes MSDCs’ accumulation and activation in TME; thus, these cells are considered a possible therapeutic target in melanoma treatment [71]. A recent study identified a set of microRNAs (miRNAs) that regulate the differentiation and polarization of MDSCs in melanoma [72]. The authors found a significant association between the levels of these circulating miRNAs and reduced PFS and OS for melanoma patients treated with PD-1 and CTLA-4 inhibitors. This relationship was not reproduced in liquid biopsies of patients treated with MAPK pathway inhibitors, indicating a possible specific correlation with resistance to immunotherapy, as opposed to targeted therapy. These data suggest the possibility that combined treatments to inhibit myeloid dysfunctions could be able to overcome resistance to ICI in melanoma [72].
As reported by Gebhardt C. and collaborators, high levels of MDSCs in the TME are associated with ipilimumab resistance. Analysis of peripheral blood of 59 metastatic melanoma patients showed increased levels of MDSCs and their chemoattractant factors in patients poorly responsive or resistant to treatment with the CTLA-4 inhibitor. In addition, MDSCs exhibited a higher production of nitric oxide and were characterized by a higher expression of PDL-1, as compared to those isolated from responsive patients [73].
In melanoma, high levels of MDSCs are also associated with resistance to BRAF inhibitors. A recent preclinical study conducted in BRAF-inhibitor resistant mouse models showed that, after an initial response to treatment, these mice develop acquired resistance associated to an increase of MDSCs in TME. MAPK signaling reactivation in BRAF-resistant mice promotes the release of a complex system of stimulating cytokines, including C-C Motif Chemokine Ligand 2 (CCL2), that attract MDSCs and suppress the immune response [74].
3.1.4. Tumor-Associated Macrophages (TAMs)
Macrophages are immune cells involved in phagocytosis, pro-inflammatory cytokines’ production, and specific immunity. The tumor-associated macrophages (TAMs), under the influence of cancer cells and the other microenvironment components, may be promoters or repressors of the tumorigenic process [75]. They are divided into two categories. M1-like macrophages (M1-TAMs), with antitumor activity, are important for the early stages of the inflammatory response. M2-like macrophages (M2-TAMs), predominant in TME, are correlated with tumor progression [76][77]. Different studies confirmed the key role of TAMs in tumor progression and have highlighted the significant correlation between the high levels of TAMs in the TME and poor prognosis for the patients [78][79]. In particular, melanoma cells, releasing miRNA-125b-5p in the microenvironment, inhibit the lysosomal acid lipase A (LIPA) and promote M2-macrophages’ phenotype and their survival [80].
TAMs induce resistance to MAPK inhibitors by favoring the expression of tumor resistance factors or through their direct paradoxical activation of the pathway, driven by BRAF inhibitors. In in vivo melanoma models, MAPK-targeted agents induce tumor necrosis factor α (TNFα) production by macrophages, which promotes NFkB pathway activation and higher MITF expression. To overcome the TNFα- and MITF-mediated resistance to MAPK inhibitors, the authors proposed a selective inhibition of NFkB pathway that, in in vitro and in vivo analyses, synergized with MEK blockade and decreased the TNFα production [81]. Moreover, TAMs suffer the paradoxical reactivation of the MAPK pathway under the influence of BRAF inhibitors, with increased production of pro-angiogenic factors, such as VEGF and IL-8 and resistance to treatments [82].
TAMs express high levels of V-domain Ig suppressor of T cell activation (VISTA), another negative immune checkpoint, which is correlated with resistance to immunotherapy [83][84]. VISTA, associated with a significant decrease of survival in primary melanomas, in vivo, promotes a protumoral microenvironment mediated by upregulation of Tregs’ levels and PDL-1 expression on macrophages’ surface [84][85][86][87].
The switch of TAMs to an antitumor phenotype is considered an alternative approach to evade TAM-mediated resistance and to reconstitute the response to PD-1 axis inhibitors. TAMs’ transformation process, mediated by STAT6 inactivation and NFkB phosphorylation, results in an increase of interleukin 12 (IL-12) production by TAMs and a reduction in inhibitory cytokine levels, such as IL-10 and C-C motif chemokine 22 (CCL22) [88]. TAMs induce immunotherapy resistance also by inhibiting the recruitment of CD8+ T lymphocytes in the tumor site. Indeed, TAMs, by stable and durable interactions with T cells, lead to the maintenance of an immunosuppressive microenvironment not responsive to the inhibitory activity of anti-PD1 molecules [89].
3.1.5. DCs
DCs are immune cells derived from myeloid precursors and are implicated in recognition and capture of antigens considered "foreign", such as pathogens or cancer cells. They are antigen presenting cells (APCs) and interact with T lymphocytes through MHC present on their surface. DCs are involved in the production of cytokines and chemokines with anti- or pro-inflammatory function according to the stimuli received from the surrounding environment [90][91][92].
Unlike other immune cells, the involvement of DC in resistance to targeted and immunotherapy is mainly associated with the absence in tumor infiltrate of this type of cell. Melanoma is able to elude the complex mechanism of T cell activation by influencing DCs’ maturation. Tumor cells produce inhibitory cytokines such as IL-8, IL-10, and VEGF and create an unfavorable environment for DCs’ maturation. This condition negatively affects the DCs’ ability to present antigen to T cells and, therefore, determines a reduced immune response [93]. Moreover, López González and collaborators demonstrated that, if inhibition of glycogen synthase kinase 3 beta (GSK3β) obstructs DC differentiation, a constitutively active GSK3β overcomes the IL-10 inhibition, leading to DC maturation [94]. In addition, melanoma promotes the switch of myeloid cells through immuno-suppressive macrophage-like cells rather than DCs [95]. The use of oncolytic virus (i.e., ORCA-010) could stimulate a specific differentiation of DCs and T cell priming by producing tumor-associated neo-antigen in order to increase the response to ICIs [96]. The therapeutic potential of DC vaccines was, recently, supported in an interesting preclinical work by Zhou and collaborators. The authors produced in vitro CD103+ murine and evaluated its activity in murine models of melanoma and osteosarcoma. CD103+ stimulated a favorable environment to the action of T lymphocytes, resulting in a reduced primary and metastatic tumor growth [97]. Further and recent results have also been obtained by direct DC targeting with molecular inhibitors. A recent study demonstrated that dasatinib (TK inhibitor) induces the activation of allogenic T cells by impairing the phosphorylation and metabolism of tryptophan induced by Indoleamine-2,3-dioxygenase (IDO), one of the most important intermediary cancer tolerants [98]. In addition, the same RAF kinase inhibitors could induce acquired resistance by influencing the differentiation and activation of DCs. Preclinical experiments, carried out on human and mouse DC cells, have detected a reduced or lack of DCs’ ability to recruit T cells after treatment with RAF kinase inhibitors. These experiments, therefore, open possible new therapeutic scenarios, not only in melanoma, considering the negative effects of pan-RAF inhibitors on the immune response modulation [99].
3.2. ECM
ECM is an intricate network of proteins, proteoglycans, and glycoconjugates produced by TME cells and involved in the adhesion and support of the cellular compartment [100]. In different cancer contexts, the tumor matrix, creating a physical barrier, blocks drugs and inhibits their action. The mutual interactions between tumor and stroma cells induce rearrangements of the matrix architecture; this remodeling promotes tumor progression and modulates response to treatment.
Fibronectin, produced by CAFs, represents the major component of ECM and seems to play a key role in a decrease of sensitivity to therapies in different solid tumors [101][102]. BRAF-mutant and PTEN-loss melanomas show, after an initial response to treatments, the development of resistance to BRAF inhibitors mediated by reactivation of MAPK and PI3K pathways. In this molecular background, drug resistance seems to be mediated also by the protective effect of fibronectin, upregulated by BRAF inhibition [103]. Phosphoproteomic analysis conducted on BRAF-mutant melanoma cell lines showed, after treatment with vemurafenib or after BRAF gene silencing, an increased expression of fibronectin, only in PTEN-loss contexts. This evidence and further experiments conducted in cells genetically manipulated for PTEN have promoted the idea that regulation of fibronectin expression is associated with PTEN. Moreover, clinical data confirmed the higher expression of fibronectin in tissue of melanoma patients with PTEN loss [103]. The interaction between fibronectin and its receptor integrin α5β1 leads to AKT phosphorylation and decreases the apoptotic capacity of melanoma cells as a result of higher activity of induced myeloid leukemia cell differentiation protein 1 (MCL-1). Therefore, BRAF inhibition promotes a remodeling of melanoma microenvironment, by which the tumor cells escape to pharmacological blockade. In the molecular contexts analyzed, the study proposed BRAF/PI3K inhibitor combinations as an alternative to overcome the development of secondary resistance to BRAF-targeted agents [103].
Integrins are transmembrane receptors that physically regulate the interaction between cells and the ECM components and promote the development of intracellular signals. To date, 24 receptors have been identified, consisting of 18 subunits of α and eight of β, and several integrins are associated with melanoma progression and metastasis [104][105]. Integrin α5β1 is the most important fibronectin receptor and their interaction modulates several cellular processes, such as adhesion, migration, and cellular differentiation [106]. An interesting work of intravital imaging of BRAF-mutant melanoma cells showed that, in co-culture systems, the treatment with BRAF inhibitors determines reactivation of MAPK pathway in areas with high stromal density; this condition is influenced by matrix remodeling associated to integrin β1/FAK/Src signaling. Under the influence of treatment, the tumor fibroblasts suffer a paradoxical activation of ERK, resulting in higher fibronectin production and interaction with its receptor on tumor cells. Integrin α5β1 promotes in melanoma cells the FAK-mediated ERK reactivation and resistance to BRAF inhibitors. Integrin αvβ3 is another receptor involved, in melanoma context, in immunotherapy resistance through its regulation of the PDL-1 expression [107]. In in vitro and in vivo melanoma models, several evidences showed that integrin αvβ3 promotes the expression of PDL-1 through activation of STAT1 [108].
The matrix remodeling is influenced by a family of metalloproteinases, enzymes involved in hydrolysis of the other proteins and in mobility of tumor cells. In melanoma, several MMPs are involved in the different aspects of tumorigenic process, such as drug resistance [109]. Vemurafenib treatment in melanoma-resistant cells supports the paradoxical reactivation of ERK, a significant increase of IL-8 levels, and activation of MMPs, especially MMP2. This condition promotes the matrix disorganization, tumor motility, and immune evasion [110].
4. Conclusions
Currently, targeted therapy and immunotherapy represent a consolidated reality for the treatment of many aggressive tumors, such as metastatic melanoma. However, like previous therapeutic approaches, they are not exempt from the problem of innate and acquired resistance, which determines the failure of treatment in an important cohort of patients. Until a few years ago, scientific research was focused on the genetic and somatic changes by which cancer cells evaded drug inhibition. However, the new evidences on TME have revealed the existence of an intricate network of interconnections between the tumor and the surrounding microenvironment that massively influences all phases of the tumorigenic process. Through direct contact or release of soluble factors, the components of TME continuously influence tumor cells’ activity modulating the drug response and therapeutic outcome. For these reasons, the research activities have focused on identifying the mechanisms and phenomena that TME implements to induce resistance to treatments. Based on the molecular mechanisms described in this review, it is evident that CAFs play a key role in the development of resistance to targeted therapy, especially through the production of many paracrine factors. Instead, the recruitment of immunosuppressive components, such as Tregs and MDSCs, in the TME is the primary mechanism of resistance to immunotherapy.
References
- Ferlay, J.; Shin, H.-R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2010, 127, 2893–2917, doi:10.1002/ijc.25516.
- Siegel, R.L.; Mph, K.D.M.; Jemal, A. Cancer statistics, 2020. CA: A Cancer J. Clin. 2020, 70, 7–30, doi:10.3322/caac.21590.
- Bennett, D.C. REVIEW ARTICLE: How to make a melanoma: what do we know of the primary clonal events? Pigment. Cell Melanoma Res. 2007, 21, 27–38, doi:10.1111/j.1755-148x.2007.00433.x.
- Shain, A.H.; Yeh, I.; Kovalyshyn, I.; Sriharan, A.; Talevich, E.; Gagnon, A.; Dummer, R.; North, J.; Pincus, L.; Ruben, B.; et al. The Genetic Evolution of Melanoma from Precursor Lesions. New Engl. J. Med. 2015, 373, 1926–1936, doi:10.1056/nejmoa1502583.
- Sanlorenzo, M.; Vujic, I.; Posch, C.; Dajee, A.; Yen, A.; Kim, S.; Ashworth, M.; Rosenblum, M.D.; Algazi, A.; Osella-Abate, S.; et al. Melanoma immunotherapy. Cancer Biol. Ther. 2014, 15, 665–74, doi:10.4161/cbt.28555.
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. New Engl. J. Med. 2017, 377, 1345–1356, doi:10.1056/nejmoa1709684.
- Dummer, R.; Ascierto, P.A.; Gogas, H.; Arance, A.; Mandalà, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsová, I.; Gutzmer, R.; et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF -mutant melanoma (COLUMBUS): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018, 19, 603–615, doi:10.1016/s1470-2045(18)30142-6.
- Long, G.V.; Flaherty, K.T.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; De Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: long-term survival and safety analysis of a phase 3 study. Ann. Oncol. 2017, 28, 1631–1639, doi:10.1093/annonc/mdx176.
- Ascierto, P.A.; Ferrucci, P.F.; Fisher, R.; Del Vecchio, M.; Atkinson, V.; Schmidt, H.; Schachter, J.; Queirolo, P.; Long, G.V.; Di Giacomo, A.M.; et al. Dabrafenib, trametinib and pembrolizumab or placebo in BRAF-mutant melanoma. Nat. Med. 2019, 25, 941–946, doi:10.1038/s41591-019-0448-9.
- Wu, T.; Dai, Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017, 387, 61–68, doi:10.1016/j.canlet.2016.01.043.
- Conciatori, F.; Bazzichetto, C.; Falcone, I.; Pilotto, S.; Bria, E.; Cognetti, F.; Milella, M.; Ciuffreda, L. Role of mTOR Signaling in Tumor Microenvironment: An Overview. Int. J. Mol. Sci. 2018, 19, 2453, doi:10.3390/ijms19082453.
- Kubo, N.; Araki, K.; Kuwano, H.; Shirabe, K. Cancer-associated fibroblasts in hepatocellular carcinoma. World J. Gastroenterol. 2016, 22, 6841–6850,, doi:10.3748/wjg.v22.i30.6841.
- Yuan, Y.; Jiang, Y.-C.; Sun, C.; Chen, Q. Role of the tumor microenvironment in tumor progression and the clinical applications (Review). Oncol. Rep. 2016, 35, 2499–2515, doi:10.3892/or.2016.4660.
- Hu, B.; Wu, Z.; Jin, H.; Hashimoto, N.; Liu, T.; Phan, S.H. CCAAT/Enhancer-Binding Protein β Isoforms and the Regulation of α-Smooth Muscle Actin Gene Expression by IL-1β. J. Immunol. 2004, 173, 4661–4668, doi:10.4049/jimmunol.173.7.4661.
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers 2015, 7, 2443–2458, doi:10.3390/cancers7040902.
- Hutchenreuther, J.; Vincent, K.; Norley, C.; Racanelli, M.; Gruber, S.B.; Johnson, T.M.; Fullen, D.R.; Raskin, L.; Perbal, B.; Holdsworth, D.W.; et al. Activation of cancer-associated fibroblasts is required for tumor neovascularization in a murine model of melanoma. Matrix Biol. 2018, 74, 52–61, doi:10.1016/j.matbio.2018.06.003.
- Cornil, I.; Theodorescu, D.; Man, S.; Herlyn, M.; Jambrosic, J.; Kerbel, R.S. Fibroblast cell interactions with human melanoma cells affect tumor cell growth as a function of tumor progression. Proc. Natl. Acad. Sci. 1991, 88, 6028–6032, doi:10.1073/pnas.88.14.6028.
- Jobe, N.P.; Rösel, D.; Dvořánková, B.; Kodet, O.; Lacina, L.; Mateu, R.; Smetana, K.; Brábek, J.; Smetana, K. Simultaneous blocking of IL-6 and IL-8 is sufficient to fully inhibit CAF-induced human melanoma cell invasiveness. Histochem. Cell Biol. 2016, 146, 205–217, doi:10.1007/s00418-016-1433-8.
- Zhou, L.; Yang, K.; Wickett, R.R.; Kadekaro, A.L.; Zhang, Y. Targeted deactivation of cancer-associated fibroblasts by β-catenin ablation suppresses melanoma growth. Tumor Biol. 2016, 37, 14235–14248, doi:10.1007/s13277-016-5293-6.
- Guo, Y.; Zhang, X.; Zeng, W.; Zhang, J.; Cai, L.; Wu, Z.; Su, J.; Xiao, Y.; Liu, N.; Tang, L.; et al. TRAF6 Activates Fibroblasts to Cancer-Associated Fibroblasts through FGF19 in Tumor Microenvironment to Benefit the Malignant Phenotype of Melanoma Cells. J. Investig. Dermatol. 2020, doi:10.1016/j.jid.2020.03.950.
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nat. Cell Biol. 2012, 487, 500–504, doi:10.1038/nature11183.
- Capparelli, C.; Rosenbaum, S.; Berger, A.C.; Aplin, A.E. Fibroblast-derived Neuregulin 1 Promotes Compensatory ErbB3 Receptor Signaling in Mutant BRAF Melanoma*. J. Biol. Chem. 2015, 290, 24267–24277, doi:10.1074/jbc.M115.657270.
- Fedorenko, I.V.; Wargo, J.A.; Flaherty, K.T.; Messina, J.L.; Smalley, K.S. BRAF Inhibition Generates a Host-Tumor Niche that Mediates Therapeutic Escape. J. Investig. Dermatol. 2015, 135, 3115–3124, doi:10.1038/jid.2015.329.
- Hirata, E.; Girotti, M.R.; Viros, A.; Hooper, S.; Spencer-Dene, B.; Matsuda, M.; Larkin, J.; Marais, R.; Sahai, E. Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin β1/FAK signaling. Cancer Cell 2015, 27, 574–88, doi:10.1016/j.ccell.2015.03.008.
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899, doi:10.1016/j.cell.2010.01.025.
- Kaur, A.; Webster, M.R.; Marchbank, K.; Behera, R.; Ndoye, A.; Kugel, C.H.; Dang, V.M.; Appleton, J.; O’Connell, M.P.; Cheng, P.; et al. sFRP2 in the aged microenvironment drives melanoma metastasis and therapy resistance. Nat. Cell Biol. 2016, 532, 250–254, doi:10.1038/nature17392.
- Takahashi, H.; Sakakura, K.; Kawabata-Iwakawa, R.; Rokudai, S.; Toyoda, M.; Nishiyama, M.; Chikamatsu, K. Immunosuppressive activity of cancer-associated fibroblasts in head and neck squamous cell carcinoma. Cancer Immunol. Immunother. 2015, 64, 1407–1417, doi:10.1007/s00262-015-1742-0.
- Zhang, A.; Qian, Y.; Ye, Z.; Chen, H.; Xie, H.; Zhou, L.; Shen, Y.; Zheng, S. Cancer-associated fibroblasts promote M2 polarization of macrophages in pancreatic ductal adenocarcinoma. Cancer Med. 2017, 6, 463–470, doi:10.1002/cam4.993.
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479.e10, doi:10.1016/j.ccell.2018.01.011.
- Ziani, L.; Ben Safta-Saadoun, T.; Gourbeix, J.; Cavalcanti, A.; Robert, C.; Favre, G.; Chouaib, S.; Thiery, J. Melanoma-associated fibroblasts decrease tumor cell susceptibility to NK cell-mediated killing through matrix-metalloproteinases secretion. Oncotarget 2017, 8, 19780–19794, doi:10.18632/oncotarget.15540.
- Li, Z.; Zhou, J.; Zhang, J.; Li, S.; Wang, H.; Du, J. Cancer-associated fibroblasts promote PD-L1 expression in mice cancer cells via secreting CXCL5. Int. J. Cancer 2019, 145, 1946–1957, doi:10.1002/ijc.32278.
- Lee, J.H.; Shklovskaya, E.; Lim, S.Y.; Carlino, M.S.; Menzies, A.M.; Stewart, A.; Pedersen, B.; Irvine, M.; Alavi, S.; Yang, J.; et al. Transcriptional downregulation of MHC class I and melanoma de- differentiation in resistance to PD-1 inhibition. Nat. Commun. 2020, 11, 1–12, doi:10.1038/s41467-020-15726-7.
- Li, G.; Satyamoorthy, K.; Herlyn, M. N-cadherin-mediated intercellular interactions promote survival and migration of melanoma cells. Cancer Res. 2001, 61, 3819–3825.
- Flach, E.H.; Rebecca, V.W.; Herlyn, M.; Smalley, K.S.; Anderson, A.R. Fibroblasts Contribute to Melanoma Tumor Growth and Drug Resistance. Mol. Pharm. 2011, 8, 2039–2049, doi:10.1021/mp200421k.
- Tiago, M.; De Oliveira, E.M.; Brohem, C.A.; Pennacchi, P.C.; Paes, R.D.; Haga, R.B.; Campa, A.; Barros, S.B.D.M.; Smalley, K.S.; Maria-Engler, S.S. Fibroblasts Protect Melanoma Cells from the Cytotoxic Effects of Doxorubicin. Tissue Eng. Part A 2014, 20, 2412–2421, doi:10.1089/ten.tea.2013.0473.
- Singer, A.; Bosselut, R. CD4⧸CD8 Coreceptors in Thymocyte Development, Selection, and Lineage Commitment: Analysis of the CD4⧸CD8 Lineage Decision. Advances in Immunology 2004, 83, 91–131, doi:10.1016/s0065-2776(04)83003-7.
- Luckheeram, R.V.; Zhou, R.; Verma, A.D.; Xia, B. CD4+T Cells: Differentiation and Functions. Clin. Dev. Immunol. 2012, 2012, 1–12, doi:10.1155/2012/925135.
- Bazzichetto, C.; Conciatori, F.; Pallocca, M.; Falcone, I.; Fanciulli, M.; Cognetti, F.; Milella, M.; Ciuffreda, L. PTEN as a Prognostic/Predictive Biomarker in Cancer: An Unfulfilled Promise? Cancers 2019, 11, 435, doi:10.3390/cancers11040435.
- Cetintas, V.B.; Batada, N.N. Is there a causal link between PTEN deficient tumors and immunosuppressive tumor microenvironment? J. Transl. Med. 2020, 18, 45–11, doi:10.1186/s12967-020-02219-w.
- Dong, Y.; Richards, J.-A.; Gupta, R.; Aung, P.P.; Emley, A.; Kluger, Y.; Dogra, S.K.; Mahalingam, M.; Wajapeyee, N. PTEN functions as a melanoma tumor suppressor by promoting host immune response. Oncogene 2013, 33, 4632–4642, doi:10.1038/onc.2013.409.
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2015, 6, 202–216, doi:10.1158/2159-8290.cd-15-0283.
- Xia, A.; Zhang, Y.; Xu, J.; Yin, T.; Lu, X.-J. T Cell Dysfunction in Cancer Immunity and Immunotherapy. Front. Immunol. 2019, 10, 1719, doi:10.3389/fimmu.2019.01719.
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen–specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 2010, 207, 2175–2186, doi:10.1084/jem.20100637.
- Chauvin, J.-M.; Pagliano, O.; Fourcade, J.; Sun, Z.; Wang, H.; Sander, C.; Kirkwood, J.M.; Chen, T.-H.T.; Maurer, M.; Korman, A.J.; et al. TIGIT and PD-1 impair tumor antigen-specific CD8⁺ T cells in melanoma patients. J. Clin. Investig. 2015, 125, 2046–58, doi:10.1172/JCI80445.
- Li, H.; Van Der Leun, A.M.; Yofe, I.; Lubling, Y.; Gelbard-Solodkin, D.; Van Akkooi, A.C.; Braber, M.V.D.; Rozeman, E.A.; Haanen, J.B.; Blank, C.U.; et al. Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell 2019, 176, 775–789.e18, doi:10.1016/j.cell.2018.11.043.
- Marshall, J.S.; Warrington, R.; Watson, W.; Kim, H.L. An introduction to immunology and immunopathology. Allergy, Asthma Clin. Immunol. 2018, 14, 49, doi:10.1186/s13223-018-0278-1.
- Somasundaram, R.; Zhang, G.; Fukunaga-Kalabis, M.; Perego, M.; Krepler, C.; Xu, X.; Wagner, C.; Hristova, D.; Zhang, J.; Tian, T.; et al. Tumor-associated B-cells induce tumor heterogeneity and therapy resistance. Nat. Commun. 2017, 8, 607, doi:10.1038/s41467-017-00452-4.
- Griss, J.; Bauer, W.; Wagner, C.; Simon, M.; Chen, M.; Grabmeier-Pfistershammer, K.; Maurer-Granofszky, M.; Roka, F.; Penz, T.; Bock, C.; et al. B cells sustain inflammation and predict response to immune checkpoint blockade in human melanoma. Nat. Commun. 2019, 10, 1–14, doi:10.1038/s41467-019-12160-2.
- Cabrita, R.; Lauss, M.; Sanna, A.; Donia, M.; Larsen, M.S.; Mitra, S.; Johansson, I.; Phung, B.; Harbst, K.; Vallon-Christersson, J.; et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020, 577, 561–565, doi:10.1038/s41586-019-1914-8.
- Amaria, R.N.; Reddy, S.M.; Tawbi, H.A.; Davies, M.A.; Ross, M.I.; Glitza, I.C.; Cormier, J.N.; Lewis, C.; Hwu, W.-J.; Hanna, E.; et al. Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nat. Med. 2018, 24, 1649–1654, doi:10.1038/s41591-018-0197-1.
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nat. Cell Biol. 2020, 577, 549–555, doi:10.1038/s41586-019-1922-8.
- Pahl, J.H.; Cerwenka, A. Tricking the balance: NK cells in anti-cancer immunity. Immunobiol. 2017, 222, 11–20, doi:10.1016/j.imbio.2015.07.012.
- Freud, A.G.; Mundy-Bosse, B.L.; Yu, J.; Caligiuri, M.A. The Broad Spectrum of Human Natural Killer Cell Diversity. Immun. 2017, 47, 820–833, doi:10.1016/j.immuni.2017.10.008.
- Cristiani, C.M.; Garofalo, C.; Passacatini, L.C.; Carbone, E. New avenues for melanoma immunotherapy: Natural Killer cells? Scand. J. Immunol. 2020, 91, e12861, doi:10.1111/sji.12861.
- López-Cobo, S.; Pieper, N.; Campos-Silva, C.; García-Cuesta, E.M.; Reyburn, H.T.; Paschen, A.; Valés-Gómez, M. Impaired NK cell recognition of vemurafenib-treated melanoma cells is overcome by simultaneous application of histone deacetylase inhibitors. OncoImmunology 2017, 7, e1392426, doi:10.1080/2162402X.2017.1392426.
- Kondělková, K.; Vokurková, D.; Krejsek, J.; Borska, L.; Fiala, Z.; Andrys, C. Regulatory T cells (Treg) and Their Roles in Immune System with Respect to Immunopathological Disorders. Acta Medica (Hradec Kralove, Czech Republic) 2010, 53, 73–77, doi:10.14712/18059694.2016.63.
- Chaudhary, B.; Elkord, E. Regulatory T Cells in the Tumor Microenvironment and Cancer Progression: Role and Therapeutic Targeting. Vaccines 2016, 4, 28, doi:10.3390/vaccines4030028.
- Ascierto, P.A.; Napolitano, M.; Celentano, E.; Simeone, E.; Gentilcore, G.; Daponte, A.; Capone, M.; Caracò, C.; Calemma, R.; Beneduce, G.; et al. Regulatory T cell frequency in patients with melanoma with different disease stage and course, and modulating effects of high-dose interferon-α 2b treatment. J. Transl. Med. 2010, 8, 76, doi:10.1186/1479-5876-8-76.
- Shang, B.; Liu, Y.; Jiang, S.-J.; Liu, Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: a systematic review and meta-analysis. Sci. Rep. 2015, 5, srep15179, doi:10.1038/srep15179.
- Leslie, C.; Bowyer, S.E.; White, A.; Grieu-Iacopetta, F.; Trevenen, M.; Iacopetta, B.; Amanuel, B.; Millward, M. FOXP3+ T regulatory lymphocytes in primary melanoma are associated with BRAF mutation but not with response to BRAF inhibitor. Pathol. 2015, 47, 557–563, doi:10.1097/pat.0000000000000314.
- Baumgartner, J.; Wilson, C.; Palmer, B.; Richter, D.; Banerjee, A.; McCarter, M. Melanoma Induces Immunosuppression by Up-Regulating FOXP3+ Regulatory T Cells. J. Surg. Res. 2007, 141, 72–77, doi:10.1016/j.jss.2007.03.053.
- Sumimoto, H.; Imabayashi, F.; Iwata, T.; Kawakami, Y. The BRAF–MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J. Exp. Med. 2006, 203, 1651–1656, doi:10.1084/jem.20051848.
- Zappasodi, R.; Budhu, S.; Hellmann, M.D.; Postow, M.A.; Senbabaoglu, Y.; Manne, S.; Gasmi, B.; Liu, C.; Zhong, H.; Li, Y.; et al. Non-conventional Inhibitory CD4+Foxp3−PD-1hi T Cells as a Biomarker of Immune Checkpoint Blockade Activity. Cancer Cell 2018, 33, 1017–1032.e7, doi:10.1016/j.ccell.2018.05.009.
- Zhang, B.; Chikuma, S.; Hori, S.; Fagarasan, S.; Honjo, T. Nonoverlapping roles of PD-1 and FoxP3 in maintaining immune tolerance in a novel autoimmune pancreatitis mouse model. Proc. Natl. Acad. Sci. 2016, 113, 8490–8495, doi:10.1073/pnas.1608873113.
- Gianchecchi, E.; Fierabracci, A. Inhibitory Receptors and Pathways of Lymphocytes: The Role of PD-1 in Treg Development and Their Involvement in Autoimmunity Onset and Cancer Progression. Front. Immunol. 2018, 9, 2374,, doi:10.3389/fimmu.2018.02374.
- Kamada, T.; Togashi, Y.; Tay, C.; Ha, D.; Sasaki, A.; Nakamura, Y.; Sato, E.; Fukuoka, S.; Tada, Y.; Tanaka, A.; et al. PD-1+ regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc. Natl. Acad. Sci. 2019, 116, 9999–10008, doi:10.1073/pnas.1822001116.
- Simeone, E.; Gentilcore, G.; Giannarelli, D.; Grimaldi, A.M.; Caracò, C.; Curvietto, M.; Esposito, A.; Paone, M.; Palla, M.; Cavalcanti, E.; et al. Immunological and biological changes during ipilimumab treatment and their potential correlation with clinical response and survival in patients with advanced melanoma. Cancer Immunol. Immunother. 2014, 63, 675–683, doi:10.1007/s00262-014-1545-8.
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti–CTLA-4 therapy against melanoma. J. Exp. Med. 2013, 210, 1695–1710, doi:10.1084/jem.20130579.
- Solito, S.; Marigo, I.; Pinton, L.; Damuzzo, V.; Mandruzzato, S.; Bronte, V. Myeloid-derived suppressor cell heterogeneity in human cancers. Ann. New York Acad. Sci. 2014, 1319, 47–65, doi:10.1111/nyas.12469.
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8, doi:10.1158/2326-6066.cir-16-0297.
- Umansky, V.; Sevko, A.; Gebhardt, C.; Utikal, J. Myeloid-derived suppressor cells in malignant melanoma. J. der Dtsch. Dermatol. Ges. 2014, 12, 1021–1027, doi:10.1111/ddg.12411.
- Huber, V.; Vallacchi, V.; Fleming, V.; Hu, X.; Cova, A.; Dugo, M.; Shahaj, E.; Sulsenti, R.; Vergani, E.; Filipazzi, P.; et al. Tumor-derived microRNAs induce myeloid suppressor cells and predict immunotherapy resistance in melanoma. J. Clin. Investig. 2018, 128, 5505–5516, doi:10.1172/jci98060.
- Gebhardt, C.; Sevko, A.; Jiang, H.; Lichtenberger, R.; Reith, M.; Tarnanidis, K.; Holland-Letz, T.; Umansky, L.; Beckhove, P.; Sucker, A.; et al. Myeloid Cells and Related Chronic Inflammatory Factors as Novel Predictive Markers in Melanoma Treatment with Ipilimumab. Clin. Cancer Res. 2015, 21, 5453–5459, doi:10.1158/1078-0432.ccr-15-0676.
- Steinberg, S.M.; Shabaneh, T.B.; Zhang, P.; Martyanov, V.; Li, Z.; Malik, B.T.; Wood, T.A.; Boni, A.; Molodtsov, A.; Angeles, C.V.; et al. Myeloid Cells That Impair Immunotherapy Are Restored in Melanomas with Acquired Resistance to BRAF Inhibitors. Cancer Res. 2017, 77, 1599–1610, doi:10.1158/0008-5472.CAN-16-1755.
- Yuan, A.; Chen, J.J.; Yang, P. Pathophysiology of Tumor-Associated Macrophages. Advances in Virus Research 2008, 45, 199–223, doi:10.1016/s0065-2423(07)00008-x.
- Yahaya, M.A.F.; Lila, M.A.M.; Ismail, S.; Zainol, M.; Afizan, N.A.R.N.M. Tumour-Associated Macrophages (TAMs) in Colon Cancer and How to Reeducate Them. J. Immunol. Res. 2019, 2019, 1–9, doi:10.1155/2019/2368249.
- Donzelli, S.; Milano, E.; Pruszko, M.; Sacconi, A.; Masciarelli, S.; Iosue, I.; Melucci, E.; Gallo, E.; Terrenato, I.; Mottolese, M.; et al. Expression of ID4 protein in breast cancer cells induces reprogramming of tumour-associated macrophages. Breast Cancer Res. 2018, 20, 59, doi:10.1186/s13058-018-0990-2.
- Liu, H.; Yang, L.; Qi, M.; Zhang, J. NFAT1 enhances the effects of tumor-associated macrophages on promoting malignant melanoma growth and metastasis. Biosci. Rep. 2018, 38, 38,, doi:10.1042/bsr20181604.
- Wanderley, C.W.; Colón, D.; Luiz, J.P.M.; Oliveira, F.F.; Viacava, P.R.; A Leite, C.; A Pereira, J.; Silva, C.M.; Silva, C.R.; Silva, R.L.; et al. Paclitaxel reduces tumor growth by reprogramming tumor-associated macrophages to an M1- profile in a TLR4-dependent manner. Cancer Res. 2018, 78, 5891–5900,, doi:10.1158/0008-5472.can-17-3480.
- Gerloff, D.; Lützkendorf, J.; Moritz, R.K.; Wersig, T.; Mäder, K.; Müller, L.P.; Sunderkötter, C. Melanoma-Derived Exosomal miR-125b-5p Educates Tumor Associated Macrophages (TAMs) by Targeting Lysosomal Acid Lipase A (LIPA). Cancers 2020, 12, 464, doi:10.3390/cancers12020464.
- Smith, M.P.; Sanchez-Laorden, B.; O’Brien, K.; Brunton, H.; Ferguson, J.; Young, H.L.; Dhomen, N.; Flaherty, K.T.; Frederick, D.T.; Cooper, Z.A.; et al. The immune microenvironment confers resistance to MAPK pathway inhibitors through macrophage-derived TNFα. Cancer Discov. 2014, 4, 1214–1229, doi:10.1158/2159-8290.CD-13-1007.
- Wang, T.; Xiao, M.; Ge, Y.; Krepler, C.; Belser, E.; Coral, A.L.; Xu, X.; Zhang, G.; Azuma, R.; Liu, Q.; et al. BRAF Inhibition Stimulates Melanoma-Associated Macrophages to Drive Tumor Growth. Clin. Cancer Res. 2015, 21, 1652–1664, doi:10.1158/1078-0432.ccr-14-1554.
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nat. Cell Biol. 2017, 545, 495–499, doi:10.1038/nature22396.
- Kuklinski, L.F.; Yan, S.; Li, Z.; Fisher, J.L.; Cheng, C.; Noelle, R.J.; Angeles, C.V.; Turk, M.J.; Ernstoff, M.S. VISTA expression on tumor-infiltrating inflammatory cells in primary cutaneous melanoma correlates with poor disease-specific survival. Cancer Immunol. Immunother. 2018, 67, 1113–1121, doi:10.1007/s00262-018-2169-1.
- Lines, J.L.; Sempere, L.F.; Wang, L.; Pantazi, E.; Mak, J.; O’Connell, S.; Ceeraz, S.; Suriawinata, A.A.; Yan, S.; Ernstoff, M.S.; et al. VISTA is an immune checkpoint molecule for human T cells. Cancer Res. 2014, 74, 1924–1932, doi:10.1158/0008-5472.CAN-13-1504.
- Kakavand, H.; A Jackett, L.; Menzies, A.M.; Gide, T.N.; Carlino, M.S.; Saw, R.P.M.; Thompson, J.F.; Wilmott, J.S.; Long, G.V.; Scolyer, R.A. Negative immune checkpoint regulation by VISTA: a mechanism of acquired resistance to anti-PD-1 therapy in metastatic melanoma patients. Mod. Pathol. 2017, 30, 1666–1676, doi:10.1038/modpathol.2017.89.
- Rosenbaum, S.R.; Knecht, M.; Mollaee, M.; Zhong, Z.; Erkes, D.A.; McCue, P.A.; Chervoneva, I.; Berger, A.C.; Lo, J.A.; Fisher, D.E.; et al. FOXD3 Regulates VISTA Expression in Melanoma. Cell Rep. 2020, 30, 510–524.e6, doi:10.1016/j.celrep.2019.12.036.
- Zhang, Y.; Wu, L.; Li, Z.; Zhang, W.; Luo, F.; Chu, Y.; Chen, G. Glycocalyx-Mimicking Nanoparticles Improve Anti-PD-L1 Cancer Immunotherapy through Reversion of Tumor-Associated Macrophages. Biomacromolecules 2018, 19, 2098–2108, doi:10.1021/acs.biomac.8b00305.
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guérin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti–PD-1 treatment. Proc. Natl. Acad. Sci. 2018, 115, E4041–E4050, doi:10.1073/pnas.1720948115.
- Klarquist, J.S.; Janssen, E.M. Melanoma-infiltrating dendritic cells. OncoImmunology 2012, 1, 1584–1593, doi:10.4161/onci.22660.
- Álvarez-Domínguez, C.; Calderón-González, R.; Terán-Navarro, H.; Salcines-Cuevas, D.; García-Castaño, A.; Freire, J.; Gómez-Román, J.; Rivera, F. Dendritic cell therapy in melanoma. Ann. Transl. Med. 2017, 5, 386, doi:10.21037/atm.2017.06.13.
- Wu, L.; Dakic, A. Development of dendritic cell system. Cell. Mol. Immunol. 2004, 1, 112–8.
- Passarelli, A.; Mannavola, F.; Stucci, L.S.; Tucci, M.; Silvestris, F. Immune system and melanoma biology: a balance between immunosurveillance and immune escape. Oncotarget 2017, 8, 106132–106142, doi:10.18632/oncotarget.22190.
- González, M.L.; Oosterhoff, D.; Lindenberg, J.J.; Milenova, I.; Lougheed, S.M.; Martiáñez, T.; Dekker, H.; Quixabeira, D.C.A.; Hangalapura, B.; Joore, J.; et al. Constitutively active GSK3β as a means to bolster dendritic cell functionality in the face of tumour-mediated immune suppression. OncoImmunology 2019, 8, e1631119–18, doi:10.1080/2162402X.2019.1631119.
- Van De Ven, R.; Lindenberg, J.J.; Oosterhoff, D.; De Gruijl, T.D. Dendritic Cell Plasticity in Tumor-Conditioned Skin: CD14+ Cells at the Cross-Roads of Immune Activation and Suppression. Front. Immunol. 2013, 4, 403,, doi:10.3389/fimmu.2013.00403.
- González, M.L.; Van De Ven, R.; De Haan, H.; Sluijs, J.V.E.V.D.; Dong, W.; Van Beusechem, V.W.; De Gruijl, T.D. Oncolytic adenovirus ORCA-010 increases the type 1 T cell stimulatory capacity of melanoma-conditioned dendritic cells. Clin. Exp. Immunol. 2020, 201, 145–160,, doi:10.1111/cei.13442.
- Zhou, Y.; Slone, N.; Chrisikos, T.T.; Kyrysyuk, O.; Babcock, R.L.; Medik, Y.B.; Li, H.S.; Kleinerman, E.S.; Watowich, S.S. Vaccine efficacy against primary and metastatic cancer with in vitro-generated CD103+conventional dendritic cells. J. Immunother. Cancer 2020, 8, e000474, doi:10.1136/jitc-2019-000474.
- Chu, C.-L.; Lee, Y.-P.; Pang, C.-Y.; Lin, H.-R.; Chen, C.-S.; Wen-Sheng, W. Tyrosine kinase inhibitors modulate dendritic cell activity via confining c-Kit signaling and tryptophan metabolism. Int. Immunopharmacol. 2020, 82, 106357, doi:10.1016/j.intimp.2020.106357.
- Riegel, K.; Schlöder, J.; Sobczak, M.; Jonuleit, H.; Thiede, B.; Schild, H.; Rajalingam, K. RAF kinases are stabilized and required for dendritic cell differentiation and function. Cell Death Differ. 2019, 27, 1300–1315, doi:10.1038/s41418-019-0416-4.
- Botti, G.; Cerrone, M.; Scognamiglio, G.; Anniciello, A.; Ascierto, P.A.; Cantile, M. Microenvironment and tumor progression of melanoma: New therapeutic prospectives. J. Immunotoxicol. 2012, 10, 235–252, doi:10.3109/1547691x.2012.723767.
- You, D.; Jung, S.P.; Jeong, Y.; Bae, S.Y.; Lee, J.E.; Kim, S. Fibronectin expression is upregulated by PI-3K/Akt activation in tamoxifen-resistant breast cancer cells. BMB Rep. 2017, 50, 615–620, doi:10.5483/bmbrep.2017.50.12.096.
- Erdogan, B.; Ao, M.; White, L.M.; Means, A.L.; Brewer, B.M.; Yang, L.; Washington, M.K.; Shi, C.; Franco, O.E.; Weaver, A.M.; et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J. Cell Biol. 2017, 216, 3799–3816, doi:10.1083/jcb.201704053.
- Fedorenko, I.V.; Abel, E.V.; Koomen, J.M.; Fang, B.; Wood, E.R.; Chen, Y.A.; Fisher, K.J.; Iyengar, S.; Dahlman, K.B.; Wargo, J.A.; et al. Fibronectin induction abrogates the BRAF inhibitor response of BRAF V600E/PTEN-null melanoma cells. Oncogene 2015, 35, 1225–35, doi:10.1038/onc.2015.188.
- Takada, Y.; Ye, X.; Simon, S. The integrins. Genome Biol. 2007, 8, 1–9, doi:10.1186/gb-2007-8-5-215.
- Huang, R.; Rofstad, E.K. Integrins as therapeutic targets in the organ-specific metastasis of human malignant melanoma. J. Exp. Clin. Cancer Res. 2018, 37, 92, doi:10.1186/s13046-018-0763-x.
- Jang, I.; Beningo, K.A. Integrins, CAFs and Mechanical Forces in the Progression of Cancer. Cancers 2019, 11, 721, doi:10.3390/cancers11050721.
- Felding-Habermann, B.; Ruggeri, Z.M.; A Cheresh, D. Distinct biological consequences of integrin alpha v beta 3-mediated melanoma cell adhesion to fibrinogen and its plasmic fragments. J. Biol. Chem. 1992, 267, 5070–5077.
- Vannini, A.; Leoni, V.; Barboni, C.; Sanapo, M.; Zaghini, A.; Malatesta, P.; Campadelli-Fiume, G.; Gianni, T. αvβ3-integrin regulates PD-L1 expression and is involved in cancer immune evasion. Proc. Natl. Acad. Sci. 2019, 116, 20141–20150, doi:10.1073/pnas.1901931116.
- Hofmann, U.B.; Westphal, J.R.; Van Muijen, G.N.; Ruiter, D.J. Matrix Metalloproteinases in Human Melanoma. J. Investig. Dermatol. 2000, 115, 337–344, doi:10.1046/j.1523-1747.2000.00068.x.
- Sandri, S.; Faião-Flores, F.; Tiago, M.; Pennacchi, P.C.; Massaro, R.R.; Alves-Fernandes, D.K.; Berardinelli, G.N.; Evangelista, A.F.; Vazquez, V.D.L.; Reis, R.M.; et al. Vemurafenib resistance increases melanoma invasiveness and modulates the tumor microenvironment by MMP-2 upregulation. Pharmacol. Res. 2016, 111, 523–533, doi:10.1016/j.phrs.2016.07.017.

