Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Maykel Hernández-Mesa | -- | 5588 | 2022-06-14 13:56:09 | | | |
| 2 | Jason Zhu | -1 word(s) | 5587 | 2022-06-15 03:23:58 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Hernández-Mesa, M.; Moreno-González, D. Mass Spectrometry in Determination of Pesticide Residues. Encyclopedia. Available online: https://encyclopedia.pub/entry/24017 (accessed on 23 July 2026).
Hernández-Mesa M, Moreno-González D. Mass Spectrometry in Determination of Pesticide Residues. Encyclopedia. Available at: https://encyclopedia.pub/entry/24017. Accessed July 23, 2026.
Hernández-Mesa, Maykel, David Moreno-González. "Mass Spectrometry in Determination of Pesticide Residues" Encyclopedia, https://encyclopedia.pub/entry/24017 (accessed July 23, 2026).
Hernández-Mesa, M., & Moreno-González, D. (2022, June 14). Mass Spectrometry in Determination of Pesticide Residues. In Encyclopedia. https://encyclopedia.pub/entry/24017
Hernández-Mesa, Maykel and David Moreno-González. "Mass Spectrometry in Determination of Pesticide Residues." Encyclopedia. Web. 14 June, 2022.
Copy Citation
The extensive use of pesticides represents a risk to human health. Consequently, legal frameworks have been established to ensure food safety, including control programs for pesticide residues. In this context, the performance of analytical methods acquires special relevance. Such methods are expected to be able to determine the largest number of compounds at trace concentration levels in complex food matrices, which represents a great analytical challenge. Technical advances in mass spectrometry (MS) have led to the development of more efficient analytical methods for the determination of pesticides.
pesticides
mass spectrometry
single-residue methods
1. Introduction
In accordance with guidance documents for the analytical quality control and method validation procedures for pesticide residue analysis, mass spectrometry (MS) is the detection technique recommended for use in pesticide analysis, as other selective detectors for gas chromatography (GC) and liquid chromatography (LC) offer limited specificity [1][2]. Despite this fact, the use of these detection systems instead of MS is possible in pesticide analysis, and can be considered in monitoring methods. However, it is essential to note that MS provides more confidence for compound identification, and has been the gold standard for pesticide determination for the last 20 years [3]. The number and type of ions that must be observed for the correct identification of the analytes of interest by MS is indicated by the SANTE/2020/12830 guideline, and varies between 2 and 3 ions when using low-resolution mass spectrometry (LRMS), or requires 2 ions with a mass accuracy lower than 5 ppm when applying high-resolution mass spectrometry (HRMS) [1].
In addition to selectivity and specificity, concentration sensitivity is one of the main analytical parameters to be taken into account in a method for the determination of pesticides. These substances are usually present in food samples at concentration levels of µg/kg, or even ng/kg. Therefore, analytical methods must provide low limits of detection (LODs) to detect such a concentration of residues. In monitoring methods for pesticide residues in food commodities, the validated LOQ is generally at the default MRL of 0.01 mg/kg [1]. In MS-based methods, the sensitivity is highly dependent on the ionization source used for LC (or GC)-MS coupling, as well as the type of mass spectrometer used. Researchers discusses the most commonly used ionization techniques in pesticide analysis when applying LC–MS or GC–MS workflows, as well as the analytical strategies followed for data acquisition, depending on the type of mass spectrometer used. In addition, new trends in MS for pesticide analysis are covered, such as ion mobility-mass spectrometry (IM–MS) hyphenation and ambient (desorption/ionization) mass spectrometry (AMS).
2. Ionization Sources
Electrospray ionization (ESI) is the most commonly applied ionization technique in LC–MS. The ESI technique is characterized by high ionization efficiency (i.e., the effectiveness of producing gas-phase ions from analytes in solution) and high transmission efficiency (i.e., the ability to transfer the ions from atmospheric pressure to the low-pressure zone of the MS system); therefore, high sensitivity is generally observed in LC–ESI-MS methods [4]. ESI is a soft ionization technique that not only provides high concentration sensitivity under properly optimized source conditions, but also often provides information on the protonated or deprotonated ions of pesticides. This information is of great relevance for the identification of unknown molecules in non-targeted analysis (e.g., pesticide transformation products), as this feature is advantageous to identify the molecular mass of the analytes. In contrast, the mass spectra resulting from ESI-MS analyses provide little information about the structure of the analytes, making structural elucidation difficult [5]. This fact also hampers the simultaneous determination of a large number of analytes in a single run [6]. Therefore, fragmentation experiments are required to overcome these problems, as well as to comply with current legislation and recommendations regarding the number of ions to be observed [1]. However, this does not represent a technical limitation at present. In-source fragmentation can be investigated to collect the ions required for identification, but can lead to complex mass spectra due to background interference that complicates the identification process. Therefore, MS/MS (or MS2) is preferred to clearly obtain sufficient characteristic fragment ions for increased confidence in pesticide identification [7], whether in targeted or non-targeted analyses.
Furthermore, although almost all current LC–MS methods for pesticide analysis involve ESI as ionization mode, other soft ionization techniques—such as atmospheric-pressure photoionization (APPI) and atmospheric pressure chemical ionization (APCI)—can also be used. ESI has been shown to be more universal than APCI for the ionization of many pesticides, whereas photoionization of pesticides has rarely been reported [8]. A large number of multiresidue methods and mega-methods have been proposed for the determination of pesticide residues by LC–ESI-MS in a wide variety of feed [9] and food products, such as vegetables and fruits [10] or honey [11].
Electron impact (EI) ionization has been the most predominant ionization technique in GC–MS. In this framework, GC–EI-MS methods were commonly applied in the control of pesticide residues in food until the widespread implementation of LC–ESI-MS methods for pesticide analysis. LC–ESI-MS has been shown to achieve higher concentration sensitivity compared to GC–EI-MS for most classes of pesticides, except for pyrethrins and organochlorines [12]. This better performance could be one of the reasons that explains the shift from GC–EI-MS to LC–ESI-MS methods for pesticide determination. In this sense, the higher sensitivity observed in the LC–ESI-MS methods may be related to the larger sample volumes that are injected into LC columns than into GC columns, and to the lower fragmentation observed in ESI sources compared to EI ionization [8].
Recently, the implementation of the APCI source in GC–MS instruments has significantly improved the sensitivity typically observed in GC–EI-MS systems [12]. For example, a 1–8-fold increase in sensitivity has been observed for the determination of organophosphorus pesticides when an APCI source is used instead of an EI source [13]. APCI is a much softer ionization technique compared to EI. Therefore, in-source fragmentation is reduced, and the protonated or deprotonated ions of the compounds can be observed in satisfactory abundance [14]. Due to the ionization mechanisms that occur in APCI (i.e., charge transfer and proton transfer), both the molecular ion (M+) and the protonated ion ([M + H]+) can be observed simultaneously. In this sense, water or methanol can be added as a modifier in the source chamber to promote the exclusive formation of protonated ions, thus increasing the sensitivity [15][16]. Currently, although it is a promising technique, GC–APCI-MS has rarely been applied to the determination of pesticides in food [17]. Among the studies focused on the analysis of pesticide residues in food by GC–APCI-MS, a screening method validated for 130 pesticides can be highlighted as an example of a multiresidue method applied to the determination of pesticides in a wide range of fruits and vegetables[18].
3. Matrix Effect
Regardless of the analytical approach, a similar drawback is observed in LC–MS and GC–MS methods—the so-called matrix effect. The matrix effect refers to the observed enhancement or suppression of the analytical signal for a specific analyte when determined in real samples, and compared to the analysis of its analytical standard. In the determination of pesticides, the matrix effect is calculated according to Equation (1). It is considered significant if it exceeds ±20% [1].
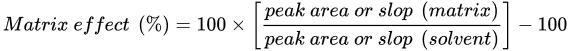
In the case of LC–MS methods, the matrix effect is associated with phenomena that occur in the ESI source. The ionization of analytes is reduced or favored by the presence of matrix components [19]. However, in the case of GC–MS methods, the matrix effect is usually caused by the retention/decomposition of analytes at the active sites (i.e., free silanol groups) in the injector, column, or detector. This is observed when standards are injected in pure solvents, but not when real samples are injected. In the latter case, matrix components can block the active sites; therefore, there are fewer active sites available to adsorb the analytes. Consequently, an enhancement in their signals is observed [20]. The determination of 341 and 315 pesticides in rice by LC–MS and GC–MS, respectively, represents a clear example of how signal suppression is usually observed in LC–ESI-MS, while signal enhancement is observed in GC–MS [21]. If the sample treatment proves ineffective in avoiding the matrix effect, it can be compensated for by applying two main strategies: (1) by using matrix-matched calibration curves (i.e., calibration curves of standards prepared in blank matrix extracts), or (2) by using isotopically labeled internal standards (IL-ISs). Both strategies are commonly applied for either LC–MS [22][23] or GC–MS methods [24][25]. It should be noted that although only one or a few IL-ISs are selected for the IL-IS approach, the quantitative performance of multiresidue methods increases when multiple IL-ISs covering different families of pesticides are used [26][27]. Other strategies to reduce the matrix effect include the possibility of applying standard addition or procedural calibration; however, matrix-matched calibration is still the preferred approach according to SANTE/2020/12830 [1]. The main challenge of using matrix-matched calibration is to find blank samples. In addition, this approach also implies the assumption that the matrix effect for a given product is the same for different samples, which is not necessarily true [28].
In GC–MS methods, the use of analyte protectants or coated inlet liners also reduces the matrix effect [20]. For example, a mixture of analyte protectants consisting of ethylglycerol, gulonolactone, D-sorbitol, and shikimic acid prepared in 4/1 (%, v/v) MeCN/water containing 0.05% (v/v) formic acid has been used for the analysis of 35 pesticides (i.e., organophosphorus pesticides, organochlorine pesticides, triazines, triazoles, carbamates, imidazoles, etc.) in tropical fruits [29]. In general, the matrix effect was significantly reduced when the analyte protectants were used. A reduction of more than three times was observed in the case of the compounds that showed a more significant matrix effect. Nevertheless, although the use of analyte protectants reduces the matrix effect and improves peak shape [30][31], these compounds must be dissolved in polar solvents such as water and MeCN. The injection of these solvents in GC systems has several drawbacks (i.e., limitations on injection volumes due to their high expansion coefficient, and poor focusing of chromatographic peaks due to their high polarity), which represents the main disadvantage of this strategy for reducing the matrix effect [20][32].
4. Targeted MS-Based Methods
Current developments in mass spectrometers and data analysis have led to a growing interest in non-targeted methods for pesticide analysis; however, targeted methods are more established in routine food testing laboratories [33]. GC–MS-based targeted methods were the technique of choice for pesticide analysis until the late 1990s [12]. GC–MS methods were initially limited to the determination of volatile and thermostable analytes, but the implementation of derivatization strategies also allowed the analysis of non-volatile compounds. In general, early GC–MS methods involved the use of a single quadrupole (Q) operating in selected ion monitoring (SIM). Nevertheless, the development of hybrid mass spectrometers made GC–triple quadrupole (QqQ)-MS, in addition to GC–Q-linear ion trap (LIT)-MS, the preferred tool for pesticide residue control. GC–MS/MS works in single reaction monitoring (SRM) or multiple reaction monitoring (MRM). This acquisition mode is very selective, because it allows monitoring of at least two fragmentation transitions per analyte (one transition corresponds to a precursor/fragment ion pair), minimizing or avoiding matrix interference, and meeting current guidelines for proper pesticide identification [34]. It is also very sensitive, because it reduces the chemical noise in the chromatogram, allowing lower LODs to be achieved [35]. Current GC–QqQ-MS-based methods allow the simultaneous determination of hundreds of pesticides, as has been shown in the analysis of 220 pesticides in sesame seeds with a separation run of 22 min [36], or in the determination of 365 pesticides in grain, beans, fruits, and vegetables with a separation time of 36 min [37].
Although LC separations have partially replaced GC separations in pesticide analysis in recent decades, QqQ-MS and QLIT-MS remain the gold standard detection tools used in analytical workflows for pesticide determination [38]. As in GC–MS/MS methods, SRM/MRM is applied as acquisition mode in LC–MS methods. Two or even three fragmentation transitions per pesticide are recorded to reduce the risk of false positives [39]. Recent multiresidue methods based on LC–MS workflows tend to cover the simultaneous analysis of a wide range of pesticides [40][41], which requires monitoring of a large number of fragmentation transitions. This is a real challenge for QqQ-MS instruments due to their slow acquisition rates. To ensure a minimum of 15 data points per chromatographic peak for satisfactory analysis reproducibility, transitions related to each analyte are only monitored within a time-limited window that covers the retention time of the compound. However, any unexpected shift in the retention time can partially or completely exclude an analyte from its detection window, resulting in truncated or undetected peaks [39]. Advanced targeted acquisition approaches for pesticide analysis should be explored to overcome this drawback following applications in other research fields [42]. For example, “Scout MRM” represents one of these advanced acquisition strategies that has already been applied to pesticide determination [39]. “Scout molecules” are compounds spiked in the samples and detected along the chromatographic separation. Once one of the “scout molecules” is detected, it triggers the monitoring of a transition group (i.e., a group of transitions from various analytes of interest). It stops upon detection of the next “scout molecule” and activation of the subsequent transition group acquisition.
5. Non-Targeted MS-Based Methods
Quadrupole instruments today are very robust, and probably provide the highest concentration sensitivity compared to other mass spectrometers, but only allow unit mass resolution. Consequently, their use is limited to targeted applications. The development of HRMS instruments has opened up new possibilities allowing semi-targeted (i.e., suspect screening) and non-targeted analysis of pesticide residues. Furthermore, high mass resolution is of great interest to avoid overestimating the amount of the target compound due to co-elution of the analyte with isobaric compounds in the matrix [43]. Depending on the HRMS technology, high mass accuracy (±0.001 Da), high mass resolution (ratio of mass to mass difference ≥ 20,000), and wide mass range (simultaneous acquisition of ions up to 2000 Da) can be obtained [44]. Mass resolving power of up to 1,000,000 can be achieved in routine analyses, specifically when using Fourier-transform ion cyclotron resonance (FT-ICR) mass spectrometers [45]. The term “resolving power” describes the ability to distinguish between ions that differ in the m/z by a small increment, and is equal to the full width at half-maximum (FWHM). FWHM is defined as m/Δm, where m represents the mass of the ion being measured, and Δm is the full width of the spectral peak at half-maximum peak height [46].
Due to the analytical performance and instrumentation cost, time-of-flight (ToF) and Orbitrap spectrometers, as well as hybrid Q-ToF and Q-Orbitrap instruments, are mainly used for pesticide determination in applications in which high mass accuracy is required [47]. More information on the capabilities of high-resolution mass spectrometers can be found in the literature [48][49]. One of the main reasons that have limited the implementation of HRMS in official pesticide controls has been the lower concentration sensitivity shown by high-resolution mass spectrometers compared to the low-resolution mass spectrometers traditionally used for this purpose. Despite QqQ-MS having been shown to be more sensitive and robust for pesticide determination at trace levels, Q-Orbitrap-MS operating in full scan can provide equal or even higher sensitivity for certain applications [43]. In fact, Orbitrap-MS has been shown to be applicable as an analytical tool in routine pesticide monitoring. Up to 210 pesticides have been determined simultaneously in fruits and vegetables under routine conditions (i.e., 102 samples), demonstrating these devices to be robust and efficient in terms of sensitivity (LOQs ≤ 10 µg/kg for the majority of pesticides and commodities) and mass accuracy (±1 mDa) [50]. Furthermore, Orbitrap-MS (140,000 FWHM, m/z 200) and ToF-MS (30,000 FWHM, m/z 556), both operating in full scan, have been compared for the quantification of pesticide residues (n = 146) in tea samples [51]. In general, similar results were observed for both mass spectrometers, but Orbitrap-MS was found to provide slightly better sensitivity and selectivity than ToF-MS at lower concentration levels (10 µg/kg), due to its higher mass resolution. Nevertheless, both mass spectrometers were demonstrated to be suitable for routine pesticide monitoring, not only because of the LODs achieved (i.e., detectable responses between 0.01 and 0.1 mg/kg, depending on the pesticide and the detector), but also in terms of intra- and inter-day precision (RSDs < 20%).
In the context of routine pesticide monitoring programs, LRMS-based methods operating in SIM or SRM/MRM have traditionally been applied to the simultaneous determination of 100–200 pesticides in a single run. However, analytical methods are evolving to cover a larger number of compounds. In this framework, HRMS-based methods have significantly expanded the testing scope of pesticides, monitoring almost 1000 analytes in less than 15 min [10]. Despite the fact that targeted analysis of pesticides can also be addressed by HRMS [52][53], HRMS-based workflows are primarily intended for non-targeted analysis of pesticides, including suspect screening approaches. The main objective of such methods is to determine pesticide residues in food products beyond a predefined scope of target pesticides. At the same time, these methods also make the identification of pesticide metabolites or (bio)transformation products feasible [54]. LC (or GC)–HRMS methods are mainly applied in two different contexts:
-
Determination of “known unknowns” (i.e., pesticides with known chemical structure and suspected presence in the food sample, but without available reference standards). Suspect screening approaches are based on screening of large lists of priority compounds using their exact mass, and subsequent tentative identification by comparing MS/MS spectra against mass spectral databases [55]. For example, Bauer et al. developed a UPLC–Q-ToF-MS method for the suspect screening of pesticide metabolites in fruit and vegetables [56]. A database of 684 metabolites of 58 active compounds was created based on relevant pesticide metabolites reported in scientific literature and approval documents. This database was subsequently applied to the tentative identification of 47 pesticide metabolites in 96 representative fruit and vegetable samples selected from a daily routine pesticide analysis. This strategy is of high interest, since the detection of metabolites in the absence of the parent pesticide may provide evidence of its illegal use.
-
Identification of “unknown unknown pesticides” without any a priori criteria, which requires a solid basic knowledge of chemistry and biochemistry to achieve an unambiguous structural elucidation of the relevant analytical signals [57]. Non-targeted analysis involves the detection of a large number of peaks, of which a significant number remain unidentified [54]. Since this analytical approach has several limitations, and does not always lead to satisfactory results to compensate for the time and effort involved in its execution, suspect screening approaches are the most commonly followed in applications related to pesticide analysis in food. In this regard, most HRMS methods for pesticide determination directly involve a list of high-interest pesticides. Consequently, they should be classified as suspect screening approaches rather than non-targeted approaches, as is often reported by many researchers. Nevertheless, great efforts are currently being made to develop suitable non-targeted methods based on HRMS to detect unexpected contaminants and chemical residues in food, such as in the case of fipronil in Dutch eggs spread in Europe in 2017, much earlier [58].
In addition, HRMS has another important advantage over LRMS. Full-scan HRMS analyses provide complete information on the content of the sample, being limited only by the satisfactory ionization and detection of the molecules under the conditions of the analytical method carried out. Therefore, raw mass spectra can be retrospectively exploited to search for a specific substance if a specific concern arises. This approach is also applicable to the determination of pesticide metabolites or (bio)transformation products whose detection was not part of the primary research objective of the original analyses. A retrospective analysis of HRMS data has been carried out for the identification of pesticides in infant milk formulas. Pesticides are not the main contaminants or chemical residues expected in this type of matrix, but they were reported to be present in 33.3% of the samples [59]. In addition, retrospective data analysis has also been carried out for the identification of pesticide transformation products in food products (i.e., honey, meat, feed, and nutraceutical products such as ginkgo, soya, green tea, and royal jelly) that were previously found to content pesticide residues. One substance—3,5,6-trichloro-2-pyridinol—was identified as transformation product of alachlor and chlorpyrifos [60]. Although retrospective analysis opens up new possibilities for wide-scope screening and reuse of raw mass data, it is not without limitations. Its application in routine analysis may be ineffective due to the necessity of high experience of the operators and the time required for compound identification, as well as for extracting information of interest from HRMS data generated without a previous hypothesis.
Most reported HRMS methods for pesticide analysis involve LC separation [47] and data acquisition in one of the following main MS modes: full scan, data-dependent acquisition (DDA), or data-independent acquisition (DIA) [61]. Full-scan acquisition involves only monitoring the accurate mass and isotopic distribution of ions reaching the detector, so full-scan data may not be sufficient for complete compound identification. Therefore, full-scan acquisition may be suitable for targeted analysis of pesticides when using HRMS [43][51], but it is not the most appropriate for suspect screening and non-targeted analysis. In these cases, product ion scan (MS2) data are needed for molecular structure identification. HRMS offers DDA and DIA modes as two data acquisition possibilities to simultaneously acquire information on the precursor ion and related fragmentation ions [62].
In DDA mode, data acquisition is carried out in full scan (MS1), and when precursor ions meet predefined criteria such as a specific m/z or an intensity threshold, they are selected as candidates for fragmentation (MS2). Conversely, in DIA mode, all precursor ions are fragmented without applying any criteria, so maximum MS2 is obtained. To date, various DIA strategies have been implemented [63], although the most commonly applied in pesticide analysis are those integrated into MS vendor software, including all-ion fragmentation (AIF) employed in Orbitrap instruments, or “all-in-one” analysis (MSE) developed by Waters. Regardless of the applied DDA or DIA mode, the collected MS2 is compared against mass spectral libraries for compound identification. DIA can potentially generate more MS2 matches, resulting in a higher chance of identifying pesticides or related compounds of interest, but without the high selectivity of DDA that provides cleaner mass spectra [61][62]. The application of DDA mode can be recommended for pesticide residue determination, since most of these compounds are known and can be effectively identified by DDA [64]. On the other hand, DIA should be implemented in those workflows aimed at the discovery of pesticide metabolites and (bio)transformation products, since these are usually unknown compounds [65]. However, recent work has shown that pesticide identification rates can be increased when the DDA and DIA modes are combined, while also providing fewer false results for the target pesticides [66][67].
Although GC–HRMS methods have been reported for the analysis of pesticides in food, they represent a lower percentage than the LC–HRMS methods developed for this purpose. GC–HRMS workflows are not yet well defined, and the possibilities of this technique have not been fully explored [12]. The commercialization of hyphenated GC–Orbitrap-MS platforms is recent (2015) and, as previously mentioned, LC–MS methods cover a wide range of applications traditionally addressed by GC–MS without requiring derivatization, and involving soft ionization (ESI vs. EI). In this sense, the ionization source for GC–HRMS coupling can also raise several questions, as occurs in GC–LRMS methods. APCI produces molecular ions that facilitate compound identification in pesticide screening [18], and is a fairly generic ionization technique for GC-amenable compounds, but is not as generic as EI [68]. In addition, generic spectral libraries for APCI-MS are still lacking, while several mass spectral libraries exist for EI-MS (e.g., NIST spectral database), which are crucial for compound identification in non-targeted analyses. Although the latter refer to nominal spectral libraries (i.e., based on EI-LRMS measurements), they are instrument-independent, and can be used for library matching if the standard ionization energy of 70 eV is used. Minor differences between EI-LRMS and EI-HRMS spectra can be observed, such as a reduced relative abundance of ions < 90 m/z when working with GC–EI-Orbitrap-MS operated in full scan compared to the NIST library [68]. However, such differences do not prevent the observation of a high match for pesticide mass spectra, thus providing high confidence in compound identification. For example, non-targeted analysis of different food commodities—such as mushrooms and peppers, among others—by GC–EI-Q-Orbitrap-MS, along with further comparison of data against libraries containing more than 200,000 spectra, allowed the detection of several pesticides (i.e., lambda-cyhalothrin, triadimenol, imazalil, pyrimethanil, and tebuconazol as the most common compounds) without any prior hypothesis [69].
6. IM–MS Hyphenation
The recent commercialization of IM–MS instruments has brought new possibilities to food analysis, and specifically to pesticide residue determination in the field of food safety [70][71]. The IMS dimension is located after the ionization source, so this analytical technique is applied to the separation of charged molecules based on their mass, charge, and shape under an electric field and in the presence of a neutral gas (e.g., typically N2, He, or CO2). In addition, chromatographic techniques (either LC or GC), ion-mobility spectrometry (IMS), and MS can be coupled, since the separation in these three dimensions occurs on different time scales (i.e., minutes/seconds, milliseconds, and microseconds, respectively). There are different IMS technologies coupled with MS on the market, including drift-tube ion-mobility spectrometry (DTIMS), traveling-wave ion-mobility spectrometry (TWIMS), trapped ion-mobility spectrometry (TIMS), high-field asymmetric-waveform ion-mobility spectrometry (FAIMS), and differential ion-mobility spectrometry (DMS). To date, pesticide determination has only been addressed by DTIMS–MS [72], TWIMS–MS [73][74][75], and DMS–MS [76]. Commercial DTIMS–MS and TWIMS–MS systems typically involve TOF mass spectrometers, because these types of analyzers offer a high acquisition rate. This instrumentation has been developed primarily for non-targeted analysis purposes. In contrast, DMS–MS systems involve QqQ-MS technology. Unlike DTIMS and TWIMS cells—where all ions entering the mobility cell are likely to reach the exit and, subsequently, be detected by MS—DMS cells act as filters of the analytes of interest [77][78]. Therefore, DMS–MS is mainly applied for targeted analysis purposes.
The high selectivity of DMS contributes to the removal of matrix compounds that co-elute in the LC dimension with target pesticides, as shown for the determination of phosphonic acid in samples that naturally contain a high concentration of phosphoric acid [79]. When applying LC–MS methods, the chromatographic separation of both substances can be compromised, while the most important qualifier mass transition of phosphonic acid is also the minor transition of phosphoric acid. Therefore, phosphoric acid can exert interference with phosphonic acid, leading to false positives. This can be avoided by integrating DMS into the LC–QqQ-MS workflow. In addition, the removal of interferences and matrix compounds in the DMS dimension not only improves method selectivity, but also improves method sensitivity by reducing chemical noise and removing adjacent chromatographic peaks from the matrix compounds. This improvement has been observed for the analysis of triazole fungicides at 0.01 mg/kg in different samples of plant origin (e.g., turnip and soy bean), which were barely identified as chromatographic peaks when applying an LC–MS method, but which could be integrated perfectly when using an LC–DMS–MS method [80].
The integration of DTMIS or TWIMS into non-targeted LC–MS workflows enhances the analytical performance of the method from three standpoints: selectivity, sensitivity, and confidence in compound identification [71]. This last aspect is of special importance for reducing the number of false positives reported in residue analysis. Both DTIMS and TWIMS (also TIMS) provide an additional identification parameter to chromatographic retention indices and mass spectra—the so-called collision cross-section (CCS). CCS is a molecular characteristic related to the spatial conformation of the ions in the gas phase. Although the drift time (i.e., the time required for ions to traverse the IMS cell) has also been used as an identification parameter in the analysis of pesticides in different fruits and vegetables [73], this parameter is instrument-dependent, and varies depending on the applied instrument settings. Nevertheless, CCS values have been shown to be reproducible between different IM–MS platforms when using the same IMS technology [81][82]. Consequently, it is possible to rely on CCS databases reported by different laboratories to use this information for identification purposes in pesticide analysis.
Several CCS databases for pesticides have been already reported [83][84]. They are also expected to be improved with CCS values for more pesticides, including transformation products and metabolites, due to the increasing implementation of LC–IM–MS instrumentation in residue analysis laboratories. In this context, the number of false positives in the screening analysis of pesticides is drastically reduced when the CCS is included in the identification criteria (i.e., threshold of ±2% from values in databases) [84]. This fact has been shown for the screening of 110 pesticides in cocoa beans at three different concentration levels (10, 50, and 150 µg/kg) [85], and for the screening of 156 pesticides at 10, 50, and 200 µg/kg in salmon feed (n = 20) [74]. In this last case, the number of false positives was reduced from 42 to 1 when the CCS was also selected as an identification criterion. Furthermore, the use of CCS values as identification criteria in the determination of pesticides by screening analysis has been shown to lead to a higher detection rate than the identification of at least one fragment ion, as this criterion is compromised for pesticides at low concentration levels [75].
The selectivity improvement provided by DTIMS or TWIMS in LC–IM–MS methods is directly related to their resolving power, which obviously depends on the IMS technology. In general, it has been estimated that the resolving power is still very limited in the LC–IM–MS instrumentation used in multiresidue analysis [77]. However, IMS has already been shown to be effective for the separation of isomeric pesticides, such as the protonated ions of ipconazole and tebufenpyrad (C18H24ClN3O)[83]. The use of current IMS technology with increased resolving power, such as cyclic-TWIMS, also allows the separation of charged isomers (e.g., protomers). This helps to understand why the relative abundance of characteristic product ions of a pesticide can vary depending on whether they are produced from analytical standards or chemical residues in food matrices [86]. Although according to SANTE 11312/2021 it is not necessary to take into account the ion ratio for pesticide identification when using HRMS, the ion ratio is a parameter to consider for this purpose when using LRMS [34]. Information on the formation of promoters for different pesticides could justify non-detected compounds present in the sample in targeted analyses when using unit mass spectrometers—due to a missing quantifier/qualifier ion, or to the alteration of their ion ratio [75]. Therefore, it could be useful to avoid false negatives when the ion ratio criterion is not fulfilled but pesticide residues are present in the sample.
Finally, DTIMS and TWIMS in LC–IM–MS workflows also isolate targeted compounds from background noise, providing cleaner mass spectra, facilitating mass spectral interpretation, and improving confidence in pesticide identification [72]. This effect also provides extracted ion chromatograms of greater quality, resulting in increased concentration sensitivity due to the improvement of the signal-to-noise ratio (S/N). Although this effect has not been extensively explored for the determination of pesticides in complex food matrices, improvements of S/N from 2- to 7-fold have been reported for the determination of small molecules when TWIMS has been integrated into an LC–MS method [87].
7. Ambient Mass Spectrometry
Ambient-ionization mass spectrometry (AIMS) is an emerging technology in the field of pesticide analysis. Compared to traditional analytical techniques used in this field, AIMS provides higher sample throughput as it involves direct, real-time, in situ analysis with minimal or no sample preparation. However, although the performance of AIMS has been studied with great interest by many routine food safety laboratories, the applications evaluated so far have not met the monitoring demands in field trials for pesticide screening. The main limitations of AIMS observed in the initial trials for routine implementation are related to the narrow analytical scope and the high rate of false positives and negatives [88]. AIMS can be considered as a promising approach for application in pesticide analysis—especially as an initial strategy to decide which samples should be sent to the laboratory for further testing. However, technical advances are still required to improve the poor reproducibility currently observed in sample analysis due to the sample heterogeneity and ion collection. These advances should also increase the concentration sensitivity, which is crucial in residue analysis but is quite limited due to the high matrix effect attributed to AIMS [89].
There is a wide range of AIMS techniques currently available for pesticide analysis, but these are mainly classified into ESI-related and APCI-related techniques. Although different AIMS approaches have been evaluated for the determination of pesticides [90], desorption electrospray ionization (DESI) and direct analysis in real time (DART) represent the most commonly used AIMS techniques in residue analysis. DESI is based on the spraying of charged solvent microdroplets at the sample surface, desorbing and ionizing the analytes that subsequently enter into the mass spectrometer [91]. The high irreproducibility of the analytical results provided by DESI is mainly associated with instrument setup and operation [92], as well as the fact that only a part of the sample surface is examined even when the pesticide residues are not homogeneously distributed on it. In this sense, the use of IL-ISs improves the reproducibility of DESI methods, and (semi)-quantification of pesticide residue levels is feasible [93]. On the other hand, DART involves the generation of plasma via the ionization of gas molecules (i.e., He, Ar, or N2), which can be consequently heated, and are impinged on the sample to cause desorption and ionization of the analytes [94]. Despite AIMS having generally been applied to the qualitative determination of pesticides in food, DART in combination with the QuPPe extraction method has been shown to be efficient for quantification purposes in the analysis of highly polar pesticides in lettuce and celery [95].
References
- SANTE/2020/12830; Guidance Document on Pesticide Analytical Methods for Risk Assessment and Post-Approval Control and Monitoring Purposes. European Commission: Brussels, Belgium, 2021; pp. 1–50.
- SANTE/12682/2019; Guidance Document on Analytical Quality Control and Method Validation for Pesticide Residues Analysis in Food and Feed. Safety of the Food Chain Pesticides and Biocides. European Commission: Brussels, Belgium, 2019; pp. 1–48.
- Parrilla Vázquez, P.; Ferrer, C.; Martínez Bueno, M.J.; Fernández-Alba, A.R. Pesticide residues in spices and herbs: Sample preparation methods and determination by chromatographic techniques. TrAC-Trends Anal. Chem. 2019, 115, 13–22.
- Lupo, S. LC-MS sensitivity: Practical strategies to boost your signal and lower your noise. LC-GC N. Am. 2018, 36, 652–661.
- De Vijlder, T.; Valkenborg, D.; Lemière, F.; Romijn, E.P.; Laukens, K.; Cuyckens, F. A tutorial in small molecule identification via electrospray ionization-mass spectrometry: The practical art of structural elucidation. Mass Spectrom. Rev. 2018, 37, 607–629.
- Fernández-Alba, A.R.; García-Reyes, J.F. Large-scale multi-residue methods for pesticides and their degradation products in food by advanced LC-MS. TrAC-Trends Anal. Chem. 2008, 27, 973–990.
- Zhang, K.; Wong, J.W.; Yang, P.; Hayward, D.G.; Sakuma, T.; Zou, Y.; Schreiber, A.; Borton, C.; Nguyen, T.V.; Kaushik, B.; et al. Protocol for an electrospray ionization tandem mass spectral product ion library: Development and application for identification of 240 pesticides in foods. Anal. Chem. 2012, 84, 5677–5684.
- Alder, L.; Greulich, K.; Kempe, G.; Vieth, B. Residue analysis of 500 high priority pesticides: Better by GC-MS or LC-MS/MS? Mass Spectrom. Rev. 2006, 25, 838–865.
- Steiner, D.; Sulyok, M.; Malachová, A.; Mueller, A.; Krska, R. Realizing the simultaneous liquid chromatography-tandem mass spectrometry based quantification of >1200 biotoxins, pesticides and veterinary drugs in complex feed. J. Chromatogr. A 2020, 1629, 461502.
- Wang, J.; Chow, W.; Wong, J.W.; Chang, J. Applications of nDATA for screening, quantitation, and identification of pesticide residues in fruits and vegetables using UHPLC/ESI Q-Orbitrap all ion fragmentation and data independent acquisition. J. Mass Spectrom. 2021, 56, e4783.
- Hrynko, I.; Łozowicka, B.; Kaczyński, P. Liquid chromatographic MS/MS analysis of a large group of insecticides in honey by modified QuEChERS. Food Anal. Methods 2018, 11, 2307–2319.
- Pico, Y.; Alfarhan, A.H.; Barcelo, D. How recent innovations in gas chromatography-mass spectrometry have improved pesticide residue determination: An alternative technique to be in your radar. TrAC-Trends Anal. Chem. 2020, 122, 115720.
- Cheng, Z.; Dong, F.; Xu, J.; Liu, X.; Wu, X.; Chen, Z.; Pan, X.; Gan, J.; Zheng, Y. Simultaneous determination of organophosphorus pesticides in fruits and vegetables using atmospheric pressure gas chromatography quadrupole-time-of-flight mass spectrometry. Food Chem. 2017, 231, 365–373.
- Fang, J.; Zhao, H.; Zhang, Y.; Lu, M.; Cai, Z. Atmospheric pressure chemical ionization in gas chromatography-mass spectrometry for the analysis of persistent organic pollutants. Trends Environ. Anal. Chem. 2020, 25, e00076.
- Portolés, T.; Mol, J.G.J.; Sancho, J.V.; Hernández, F. Advantages of atmospheric pressure chemical ionization in gas chromatography tandem mass spectrometry: Pyrethroid insecticides as a case study. Anal. Chem. 2012, 84, 9802–9810.
- Cha, K.H.; Lee, J.; Lee, J.; Kim, J.H. Development of a quantitative screening method for pesticide multiresidues in orange, chili pepper, and brown rice using gas chromatography-quadrupole time of flight mass spectrometry with dopant-assisted atmospheric pressure chemical ionization. Food Chem. 2022, 374, 131626.
- Saito-Shida, S.; Nagata, M.; Nemoto, S.; Akiyama, H. Quantitative analysis of pesticide residues in tea by gas chromatography–tandem mass spectrometry with atmospheric pressure chemical ionization. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2020, 1143, 122057.
- Cervera, M.I.; Portolés, T.; López, F.J.; Beltrán, J.; Hernández, F. Screening and quantification of pesticide residues in fruits and vegetables making use of gas chromatography-quadrupole time-of-flight mass spectrometry with atmospheric pressure chemical ionization. Anal. Bioanal. Chem. 2014, 406, 6843–6855.
- Rigano, F.; Tranchida, P.Q.; Dugo, P.; Mondello, L. High-performance liquid chromatography combined with electron ionization mass spectrometry: A review. TrAC-Trends Anal. Chem. 2019, 118, 112–122.
- Rutkowska, E.; Łozowicka, B.; Kaczyński, P. Three approaches to minimize matrix effects in residue analysis of multiclass pesticides in dried complex matrices using gas chromatography tandem mass spectrometry. Food Chem. 2019, 279, 20–29.
- Tran-Lam, T.-T.; Bui, M.Q.; Nguyen, H.Q.; Dao, Y.H.; Le, G.T. A combination of chromatography with tandem mass spectrometry systems (UPLC-MS/MS and GC-MS/MS), modified QuEChERS extraction and mixed-mode SPE clean-up method for the analysis of 656 pesticide residues in rice. Foods 2021, 10, 2455.
- Herrera López, S.; Scholten, J.; Kiedrowska, B.; de Kok, A. Method validation and application of a selective multiresidue analysis of highly polar pesticides in food matrices using hydrophilic interaction liquid chromatography and mass spectrometry. J. Chromatogr. A 2019, 1594, 93–104.
- Song, N.E.; Lee, J.Y.; Mansur, A.R.; Jang, H.W.; Lim, M.C.; Lee, Y.; Yoo, M.; Nam, T.G. Determination of 60 pesticides in hen eggs using the QuEChERS procedure followed by LC-MS/MS and GC-MS/MS. Food Chem. 2019, 298, 125050.
- Giacinti, G.; Raynaud, C.; Capblancq, S.; Simon, V. Matrix-matching as an improvement strategy for the detection of pesticide residues. J. Food Sci. 2016, 81, T1342–T1350.
- Tsuchiyama, T.; Katsuhara, M.; Sugiura, J.; Nakajima, M.; Yamamoto, A. Combined use of a modifier gas generator, analyte protectants and multiple internal standards for effective and robust compensation of matrix effects in gas chromatographic analysis of pesticides. J. Chromatogr. A 2019, 1589, 122–133.
- Tsuchiyama, T.; Katsuhara, M.; Nakajima, M. Compensation of matrix effects in gas chromatography–mass spectrometry analysis of pesticides using a combination of matrix matching and multiple isotopically labeled internal standards. J. Chromatogr. A 2017, 1524, 233–245.
- Colazzo, M.; Alonso, B.; Ernst, F.; Cesio, M.V.; Perez-Parada, A.; Heinzen, H.; Pareja, L. Determination of multiclass, semi-polar pesticide residues in fatty fish muscle tissue by gas and liquid chromatography mass spectrometry. MethodsX 2019, 6, 929–937.
- Fabregat-Cabello, N.; Zomer, P.; Sancho, J.V.; Roig-Navarro, A.F.; Mol, H.G.J. Comparison of approaches to deal with matrix effects in LC-MS/MS based determinations of mycotoxins in food and feed. World Mycotoxin J. 2016, 9, 149–161.
- Varela-Martínez, D.A.; González-Curbelo, M.Á.; González-Sálamo, J.; Hernández-Borges, J. High-throughput analysis of pesticides in minor tropical fruits from Colombia. Food Chem. 2019, 280, 221–230.
- Anastassiades, M.; Maštovská, K.; Lehotay, S.J. Evaluation of analyte protectants to improve gas chromatographic analysis of pesticides. J. Chromatogr. A 2003, 1015, 163–184.
- Soliman, M.; Khorshid, M.A.; Abo-Aly, M.M. Combination of analyte protectants and sandwich injection to compensate for matrix effect of pesticides residue in GC–MS/MS. Microchem. J. 2020, 156, 104852.
- Rutkowska, E.; Łozowicka, B.; Kaczyński, P. Compensation of matrix effects in seed matrices followed by gas chromatography-tandem mass spectrometry analysis of pesticide residues. J. Chromatogr. A 2020, 1614, 460738.
- Wong, J.W.; Wang, J.; Chow, W.; Carlson, R.; Jia, Z.; Zhang, K.; Hayward, D.G.; Chang, J.S. Perspectives on Liquid Chromatography—High-Resolution Mass Spectrometry for Pesticide Screening in Foods. J. Agric. Food Chem. 2018, 66, 9573–9581.
- Sante/11312/2021; Guidance Document on Analytical Quality Control and Method Validation for Pesticide Residues Analysis in Food and Feed. European Commission: Brussels, Belgium, 2021; pp. 1–57.
- Grimalt, S.; Dehouck, P. Review of analytical methods for the determination of pesticide residues in grapes. J. Chromatogr. A 2016, 1433, 1–23.
- Shinde, R.; Pardeshi, A.; Dhanshetty, M.; Anastassiades, M.; Banerjee, K. Development and validation of an analytical method for the multiresidue analysis of pesticides in sesame seeds using liquid- and gas chromatography with tandem mass spectrometry. J. Chromatogr. A 2021, 1652, 462346.
- Kang, H.S.; Kim, M.K.; Kim, E.J. High-throughput simultaneous analysis of multiple pesticides in grain, fruit, and vegetables by GC-MS/MS. Food Addit. Contam.-Part A Chem. Anal. Control Expo. Risk Assess. 2020, 37, 963–972.
- Stachniuk, A.; Fornal, E. Liquid chromatography-mass spectrometry in the analysis of pesticide residues in food. Food Anal. Methods 2016, 9, 1654–1665.
- Salvador, A.; Carrière, R.; Ayciriex, S.; Margoum, C.; Leblanc, Y.; Lemoine, J. Scout-multiple reaction monitoring: A liquid chromatography tandem mass spectrometry approach for multi-residue pesticide analysis without time scheduling. J. Chromatogr. A 2020, 1621, 461046.
- Kiljanek, T.; Niewiadowska, A.; Małysiak, M.; Posyniak, A. Miniaturized multiresidue method for determination of 267 pesticides, their metabolites and polychlorinated biphenyls in low mass beebread samples by liquid and gas chromatography coupled with tandem mass spectrometry. Talanta 2021, 235, 122721.
- Végh, R.; Sörös, C.; Majercsik, N.; Sipos, L. Determination of Pesticides in Bee Pollen: Validation of a multiresidue high-performance liquid chromatography-mass spectrometry/mass spectrometry method and testing pollen samples of selected botanical origin. J. Agric. Food Chem. 2022, 70, 1507–1515.
- Van Bentum, M.; Selbach, M. An introduction to advanced targeted acquisition methods. Mol. Cell. Proteom. 2021, 20, 100165.
- Belarbi, S.; Vivier, M.; Zaghouani, W.; De Sloovere, A.; Agasse-Peulon, V.; Cardinael, P. Comparison of new approach of GC-HRMS (Q-Orbitrap) to GC–MS/MS (triple-quadrupole) in analyzing the pesticide residues and contaminants in complex food matrices. Food Chem. 2021, 359, 129932.
- Hollender, J.; Schymanski, E.L.; Singer, H.P.; Ferguson, P.L. Nontarget screening with high resolution mass spectrometry in the environment: Ready to go? Environ. Sci. Technol. 2017, 51, 11505–11512.
- Bowman, A.P.; Blakney, G.T.; Hendrickson, C.L.; Ellis, S.R.; Heeren, R.M.A.; Smith, D.F. Ultra-high mass resolving power, mass accuracy, and dynamic range MALDI mass spectrometry imaging by 21-T FT-ICR MS. Anal. Chem. 2020, 92, 3133–3142.
- Urban, J.; Afseth, N.K.; Štys, D. Fundamental definitions and confusions in mass spectrometry about mass assignment, centroiding and resolution. TrAC-Trends Anal. Chem. 2014, 53, 126–136.
- Masiá, A.; Suarez-Varela, M.M.; Llopis-Gonzalez, A.; Picó, Y. Determination of pesticides and veterinary drug residues in food by liquid chromatography-mass spectrometry: A review. Anal. Chim. Acta 2016, 936, 40–61.
- Špánik, I.; Machyňáková, A. Recent applications of gas chromatography with high-resolution mass spectrometry. J. Sep. Sci. 2018, 41, 163–179.
- García-Reyes, J.F.; Moreno-González, D.; Nortes-Méndez, R.; Gilbert-López, B.; Molina Díaz, A. HRMS: Hardware and Software. In Applications in High Resolution Mass Spectrometry; Romero-González, R., Garrido, A., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; ISBN 9780128096482.
- Uclés, S.; Uclés, A.; Lozano, A.; Martínez Bueno, M.J.; Fernández-Alba, A.R. Shifting the paradigm in gas chromatography mass spectrometry pesticide analysis using high resolution accurate mass spectrometry. J. Chromatogr. A 2017, 1501, 107–116.
- Saito-Shida, S.; Hamasaka, T.; Nemoto, S.; Akiyama, H. Multiresidue determination of pesticides in tea by liquid chromatography-high-resolution mass spectrometry: Comparison between Orbitrap and time-of-flight mass analyzers. Food Chem. 2018, 256, 140–148.
- Manzano-Sánchez, L.; Martínez-Martínez, J.A.; Domínguez, I.; Vidal, J.L.M.; Frenich, A.G.; Romero-González, R. Development and application of a novel pluri-residue method to determine polar pesticides in fruits and vegetables through liquid chromatography high resolution mass spectrometry. Foods 2020, 9, 553.
- Pérez-Ortega, P.; Lara-Ortega, F.J.; Gilbert-López, B.; Moreno-González, D.; García-Reyes, J.F.; Molina-Díaz, A. Screening of over 600 pesticides, veterinary drugs, food-packaging contaminants, mycotoxins, and other chemicals in food by ultra-high performance liquid chromatography quadrupole time-of-flight mass spectrometry (UHPLC-QTOFMS). Food Anal. Methods 2017, 10, 1216–1244.
- Guo, Z.; Zhu, Z.; Huang, S.; Wang, J. Non-targeted screening of pesticides for food analysis using liquid chromatography high-resolution mass spectrometry-a review. Food Addit. Contam.-Part A Chem. Anal. Control Expo. Risk Assess. 2020, 37, 1180–1201.
- Hollender, J.; van Bavel, B.; Dulio, V.; Farmen, E.; Furtmann, K.; Koschorreck, J.; Kunkel, U.; Krauss, M.; Munthe, J.; Schlabach, M.; et al. High resolution mass spectrometry-based non-target screening can support regulatory environmental monitoring and chemicals management. Environ. Sci. Eur. 2019, 31, 42.
- Bauer, A.; Luetjohann, J.; Rohn, S.; Jantzen, E.; Kuballa, J. Development of a suspect screening strategy for pesticide metabolites in fruit and vegetables by UPLC-Q-Tof-MS. Food Anal. Methods 2018, 11, 1591–1607.
- Pourchet, M.; Debrauwer, L.; Klanova, J.; Price, E.J.; Covaci, A.; Caballero-Casero, N.; Oberacher, H.; Lamoree, M.; Damont, A.; Fenaille, F.; et al. Suspect and non-targeted screening of chemicals of emerging concern for human biomonitoring, environmental health studies and support to risk assessment: From promises to challenges and harmonisation issues. Environ. Int. 2020, 139, 105545.
- Kunzelmann, M.; Winter, M.; Åberg, M.; Hellenäs, K.E.; Rosén, J. Non-targeted analysis of unexpected food contaminants using LC-HRMS. Anal. Bioanal. Chem. 2018, 410, 5593–5602.
- Izzo, L.; Narváez, A.; Castaldo, L.; Gaspari, A.; Rodríguez-Carrasco, Y.; Grosso, M.; Ritieni, A. Multiclass and multi-residue screening of mycotoxins, pharmacologically active substances, and pesticides in infant milk formulas through ultra-high-performance liquid chromatography coupled with high-resolution mass spectrometry analysis. J. Dairy Sci. 2022, 105, 2948–2962.
- Gómez-Pérez, M.L.; Romero-González, R.; Vidal, J.L.M.; Frenich, A.G. Identification of transformation products of pesticides and veterinary drugs in food and related matrices: Use of retrospective analysis. J. Chromatogr. A 2015, 1389, 133–138.
- Fisher, C.M.; Croley, T.R.; Knolhoff, A.M. Data processing strategies for non-targeted analysis of foods using liquid chromatography/high-resolution mass spectrometry. TrAC-Trends Anal. Chem. 2021, 136, 116188.
- Li, D.; Liang, W.; Feng, X.; Ruan, T.; Jiang, G. Recent advances in data-mining techniques for measuring transformation products by high-resolution mass spectrometry. TrAC-Trends Anal. Chem. 2021, 143, 116409.
- Zhang, F.; Ge, W.; Ruan, G.; Cai, X.; Guo, T. Data-Independent Acquisition mass spectrometry-based proteomics and software tools: A glimpse in 2020. Proteomics 2020, 20, 1900276.
- Zhou, H.; Cao, Y.M.; Miao, S.; Lan, L.; Chen, M.; Li, W.T.; Mao, X.H.; Ji, S. Qualitative screening and quantitative determination of 569 pesticide residues in honeysuckle using ultrahigh-performance liquid chromatography coupled to quadrupole-Orbitrap high resolution mass spectrometry. J. Chromatogr. A 2019, 1606, 460374.
- Moschet, C.; Lew, B.M.; Hasenbein, S.; Anumol, T.; Young, T.M. LC- and GC-QTOF-MS as complementary tools for a comprehensive micropollutant analysis in aquatic systems. Environ. Sci. Technol. 2017, 51, 1553–1561.
- Rajski, Ł.; Petromelidou, S.; Díaz-Galiano, F.J.; Ferrer, C.; Fernández-Alba, A.R. Improving the simultaneous target and non-target analysis LC-amenable pesticide residues using high speed Orbitrap mass spectrometry with combined multiple acquisition modes. Talanta 2021, 228, 122241.
- Sun, F.; Tan, H.; Li, Y.; De Boevre, M.; Zhang, H.; Zhou, J.; Li, Y.; Yang, S. An integrated data-dependent and data-independent acquisition method for hazardous compounds screening in foods using a single UHPLC-Q-Orbitrap run. J. Hazard. Mater. 2021, 401, 123266.
- Mol, H.G.J.; Tienstra, M.; Zomer, P. Evaluation of gas chromatography-electron ionization-full scan high resolution Orbitrap mass spectrometry for pesticide residue analysis. Anal. Chim. Acta 2016, 935, 161–172.
- Vargas-Pérez, M.; Domínguez, I.; González, F.J.E.; Frenich, A.G. Application of full scan gas chromatography high resolution mass spectrometry data to quantify targeted-pesticide residues and to screen for additional substances of concern in fresh-food commodities. J. Chromatogr. A 2020, 1622, 461118.
- Hernández-Mesa, M.; Escourrou, A.; Monteau, F.; Le Bizec, B.; Dervilly-Pinel, G. Current applications and perspectives of ion mobility spectrometry to answer chemical food safety issues. TrAC-Trends Anal. Chem. 2017, 94, 39–53.
- Hernández-Mesa, M.; Ropartz, D.; García-Campaña, A.M.; Rogniaux, H.; Dervilly-Pinel, G.; Le Bizec, B. Ion mobility spectrometry in food analysis: Principles, current applications and future trends. Molecules 2019, 24, 2706.
- Chen, X.P.; Zhang, F.; Guo, Y.L. Validating an ion mobility spectrometry-quadrupole time of flight mass spectrometry method for high-throughput pesticide screening. Analyst 2019, 144, 4835–4840.
- Goscinny, S.; Joly, L.; De Pauw, E.; Hanot, V.; Eppe, G. Travelling-wave ion mobility time-of-flight mass spectrometry as an alternative strategy for screening of multi-class pesticides in fruits and vegetables. J. Chromatogr. A 2015, 1405, 85–93.
- Regueiro, J.; Negreira, N.; Hannisdal, R.; Berntssen, M.H.G. Targeted approach for qualitative screening of pesticides in salmon feed by liquid chromatography coupled to traveling-wave ion mobility/quadrupole time-of-flight mass spectrometry. Food Control 2017, 78, 116–125.
- Bauer, A.; Kuballa, J.; Rohn, S.; Jantzen, E.; Luetjohann, J. Evaluation and validation of an ion mobility quadrupole time-of-flight mass spectrometry pesticide screening approach. J. Sep. Sci. 2018, 41, 2178–2187.
- Kolberg, D.I.S.; Zechmann, S.; Wildgrube, C.; Sigalov, I.; Scherbaum, E.; Anastassiades, M. Determination of triazole derivative metabolites (TDMs) in fruit and vegetables using the QuPPe method and differential mobility spectrometry (DMS) and survey of the residue situation in organic and conventional produce. Asp. Food Control Anim. Health 2016, 2, 2196–3460.
- Kaufmann, A. The use of UHPLC, IMS, and HRMS in multiresidue analytical methods: A critical review. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2020, 1158, 122369.
- Schneider, B.B.; Nazarov, E.G.; Londry, F.; Vouros, P.; Covey, T.R. Differential mobility spectrometry/mass spectrometry history, theory, design optimization, simulations, and applications. Mass Spectrom. Rev. 2016, 35, 687–737.
- Anastassiades, M.; Kolberg, D.I.; Eichhorn, E.; Wachtler, A.-K.; Benkenstein, A.; Zechmann, S.; Mack, D.; Wildgrube, C.; Barth, A.; Sigalov, I.; et al. Quick method for the analysis of numerous highly polar pesticides in food involving extraction with acidified methanol and LC-MS/MS measurement. I. Food of plant origin (QuPPe-PO-Method) Version. Eurl-Srm 2020, 11, 1–101.
- Jasak, J.; Le Blanc, Y.; Speer, K.; Billian, P.; Schoening, R.M. Analysis of triazole-based metabolites in plant materials using differential mobility spectrometry to improve LC/MS/MS selectivity. J. AOAC Int. 2012, 95, 1768–1776.
- Stow, S.M.; Causon, T.J.; Zheng, X.; Kurulugama, R.T.; Mairinger, T.; May, J.C.; Rennie, E.E.; Baker, E.S.; Smith, R.D.; McLean, J.A.; et al. An interlaboratory evaluation of drift tube ion mobility-mass spectrometry collision cross section measurements. Anal. Chem. 2017, 89, 9048–9055.
- Hernández-Mesa, M.; D’Atri, V.; Barknowitz, G.; Fanuel, M.; Pezzatti, J.; Dreolin, N.; Ropartz, D.; Monteau, F.; Vigneau, E.; Rudaz, S.; et al. Interlaboratory and interplatform study of steroids collision cross section by traveling wave ion mobility spectrometry. Anal. Chem. 2020, 92, 5013–5022.
- Regueiro, J.; Negreira, N.; Berntssen, M.H.G. Ion-mobility-derived collision cross section as an additional identification point for multiresidue screening of pesticides in fish feed. Anal. Chem. 2016, 88, 11169–11177.
- Goscinny, S.; McCullagh, M.; Far, J.; De Pauw, E.; Eppe, G. Towards the use of ion mobility mass spectrometry derivedcollision cross section as a screening approach for unambiguousidentification of targeted pesticides in food. Rapid Commun. Mass Spectrom. 2019, 33, 34–48.
- Zainudin, B.H.; Salleh, S.; Yaakob, A.S.; Mohamed, R. Comprehensive strategy for pesticide residue analysis in cocoa beans through qualitative and quantitative approach. Food Chem. 2022, 368, 130778.
- McCullagh, M.; Goscinny, S.; Palmer, M.; Ujma, J. Investigations into pesticide charge site isomers using conventional IM and cIM systems. Talanta 2021, 234, 122604.
- Hernández-Mesa, M.; Monteau, F.; Le Bizec, B.; Dervilly-Pinel, G. Potential of ion mobility-mass spectrometry for both targeted and non-targeted analysis of phase II steroid metabolites in urine. Anal. Chim. Acta X 2019, 1, 100006.
- Lehotay, S.J.; Chen, Y. Hits and misses in research trends to monitor contaminants in foods. Anal. Bioanal. Chem. 2018, 410, 5331–5351.
- Hernández-Mesa, M.; Lara, F.J.; Moreno-González, D.; Olmo-Iruela, M.; García-Campaña, A.M. Trends in Multiresidue Analysis; Meyers, R.A., Ed.; Wiley: Hoboken, NJ, USA, 2020; ISBN 9780470027318.
- Beneito-Cambra, M.; Gilbert-López, B.; Moreno-González, D.; Bouza, M.; Franzke, J.; García-Reyes, J.F.; Molina-Díaz, A. Ambient (desorption/ionization) mass spectrometry methods for pesticide testing in food: A review. Anal. Methods 2020, 12, 4831–4852.
- Gentili, A.; Fanali, S.; Mainero Rocca, L. Desorption electrospray ionization mass spectrometry for food analysis. TrAC-Trends Anal. Chem. 2019, 115, 162–173.
- Shelley, J.T.; Badal, S.P.; Engelhard, C.; Hayen, H. Ambient desorption/ionization mass spectrometry: Evolution from rapid qualitative screening to accurate quantification tool. Anal. Bioanal. Chem. 2018, 410, 4061–4076.
- Gerbig, S.; Stern, G.; Brunn, H.E.; Düring, R.A.; Spengler, B.; Schulz, S. Method development towards qualitative and semi-quantitative analysis of multiple pesticides from food surfaces and extracts by desorption electrospray ionization mass spectrometry as a preselective tool for food control. Anal. Bioanal. Chem. 2017, 409, 2107–2117.
- Hajslova, J.; Cajka, T.; Vaclavik, L. Challenging applications offered by direct analysis in real time (DART) in food-quality and safety analysis. TrAC-Trends Anal. Chem. 2011, 30, 204–218.
- Lara, F.J.; Chan, D.; Dickinson, M.; Lloyd, A.S.; Adams, S.J. Evaluation of direct analysis in real time for the determination of highly polar pesticides in lettuce and celery using modified Quick Polar Pesticides Extraction method. J. Chromatogr. A 2017, 1496, 37–44.
More
Information
Subjects:
Chemistry, Analytical
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.6K
Revisions:
2 times
(View History)
Update Date:
15 Jun 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No