Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jean-Philippe Berteau | -- | 1472 | 2022-06-13 08:53:33 |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Berteau, J. Pathogenesis and Risk Factors of Osteoarthritis. Encyclopedia. Available online: https://encyclopedia.pub/entry/23957 (accessed on 24 July 2026).
Berteau J. Pathogenesis and Risk Factors of Osteoarthritis. Encyclopedia. Available at: https://encyclopedia.pub/entry/23957. Accessed July 24, 2026.
Berteau, Jean-Philippe. "Pathogenesis and Risk Factors of Osteoarthritis" Encyclopedia, https://encyclopedia.pub/entry/23957 (accessed July 24, 2026).
Berteau, J. (2022, June 13). Pathogenesis and Risk Factors of Osteoarthritis. In Encyclopedia. https://encyclopedia.pub/entry/23957
Berteau, Jean-Philippe. "Pathogenesis and Risk Factors of Osteoarthritis." Encyclopedia. Web. 13 June, 2022.
Copy Citation
Knee osteoarthritis (OA) is a degenerative joint disease and the most common reason for knee joint replacements in the US, with 4.7 million individuals having undergone surgery in 2010 with an associated cost of USD 29,488 Per surgery. The high prevalence of knee OA manifests in enormous societal and personal expenses and urges to prevent OA progression to avoid surgery.
knee pain
osteoarthritis
1. Pathogenesis
First, OA has been depicted as the result of progressive articular cartilage degradation. Indeed, although the cartilage can prevent biomechanical damage caused by severe loading, patients with OA hinder attempts at repair and result in disrupted cartilage homeostasis [1]. For instance, cartilage cells’ (i.e., chondrocytes’) compositional and structural alterations—such as hypertrophy due to aging or oxidative stress—trigger the production of catabolic factors, enhancing cartilage debilitation. These catabolic factors such as cytokines, chemokines, and proteolytic enzymes—cytokines (e.g., IL-6, IL-8), chemokines (e.g., RANTES, IP-10), metalloproteases (MMP1, MMP3), and heat-shock proteins (e.g., HSPA1A)—have been identified as quantifiable biomarkers for predicting the onset and progression of knee OA. Therefore, for decades, cartilage degradation resulting from the extracellular matrix’s destruction has been depicted as one of the significant biological starters of the OA pathological process.
When the pathological process of knee OA is triggered by catabolic factors production, the articular cartilage of the knee starts to degrade, making it unable to fully absorb physiological and physical forces. This induces associated joint conformational changes that compensate for the loss of articular cartilage, showing that OA is an active repair process [2]. These changes include subchondral bone (SB) sclerosis—thickening and hardening—and the formation of bone cysts and marginal osteophytes (coming from bone remodeling). Thus, all these SB alterations cause the joint space to narrow [3], enhancing OA progression. Ultimately, OA affects the whole joint due to synovial inflammation and fibrosis of the joint capsule [4], which cause loss of range of motion/stiffness, tenderness, and pain. This pathological process has been described in the shape of a vicious circle of OA when one event triggers the other [3].
Regarding pain in OA disease, it involves complex peripheral and central mechanisms. For instance, nerve sensitizations are significant characteristics of pain transmission in OA patients that may contribute to the discordance between pain and joint pathology [5]. Since hyaline cartilage is not innervated, the pain comes from the synovium, subchondral bone, and periosteum, which are innervated by small-diameter nociceptive neurons. The nociceptive stimuli are generated by tissue damage during joint degradation. Previous studies showed pain had been associated with many structural factors, including bone marrow lesions, synovial thickening (synovitis), and knee effusion [6]. The inflammatory mediators produced by the synovium and chondrocytes increase the excitation of the nociceptive neurons, creating an amplified painful response [7]. Recent evidence shows that SB is also a starter of the OA vicious circle. Indeed, OA is also looked at as joint failure caused by abnormal joint loading instead of a disease of cartilage degradation [8]. Specifically, changes in the SB—which can trigger pain through nociceptive stimuli—predispose the cartilage to further damage from wear and tear, as the SB is less able to absorb forces/load placed on the joint [9][10][11][12][13][14]. It is believed that changes to the mechanical properties of SB occur during remodeling, such as bone hardening [13], which induces increased stiffness that precedes and contributes to cartilage loss. Indeed, it has been shown that changes in gene expression of SB precede cartilage degeneration and alter the activity of catabolic factors by chondrocytes contributing to the degeneration of cartilage [12]. Thus, alterations of physiological cross-talk between SB and cartilage [15] are considered the primary trigger of the OA pathological process. OA is a self-sustaining vicious cycle (adapted from [3]) where each step in the process influences and amplifies each other (Figure 1).
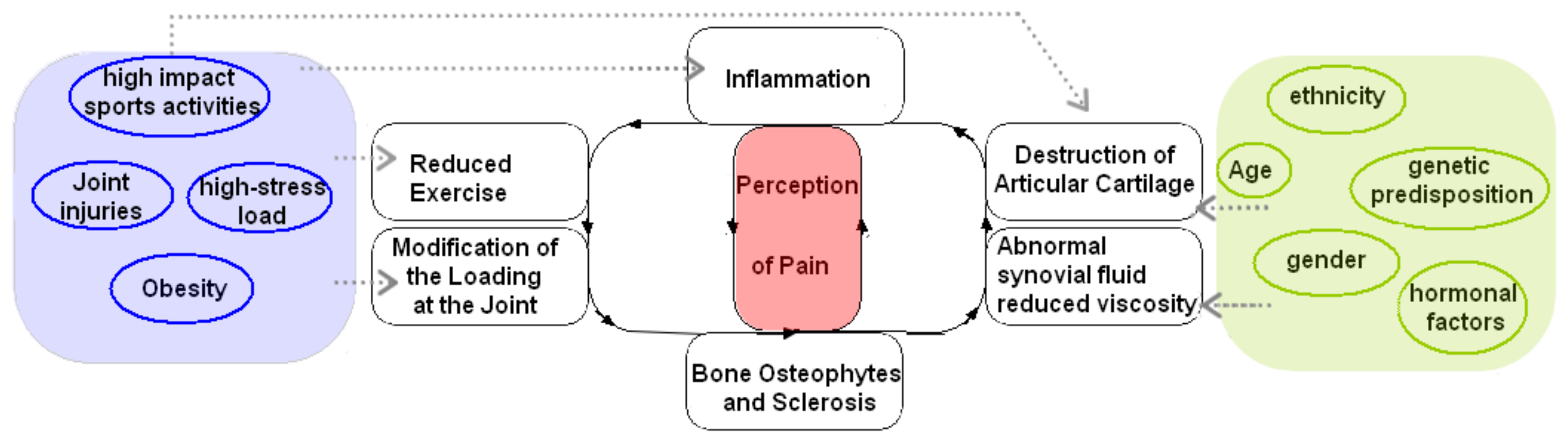
Figure 1. The vicious circle of OA progression and risk factors are associated where pain perception is central to the disease.
2. Risk Factors
The development of the vicious cycle of OA is complex and is caused by both modifiable and nonmodifiable risk factors. Indeed, no one risk factor contributes to the increase in the disease process; rather, the involvement of risk factors together such as age, gender, ethnicity, genetic predisposition, hormonal factors, and bone density. In addition, biomechanical factors—caused by sports, the workplace, joint misalignment, and obesity—contribute to joint injuries leading to OA [3][7][16][17].
2.1. Age
There is an exponential increase in OA in adults over 50 years old. Indeed, the aging process results in the chondrocytes’ inability to produce proteoglycans to maintain the cartilage matrix—which gives the cartilage its compressive strength—and the failure to maintain homeostasis [7]. Thus, the tissue is less likely to heal when stressed, causing articular cartilage degeneration, leading to OA. However, it cannot be a purely age-related joint wear and tear disease because not all joints are equally affected, and OA changes can develop without aging [7]. For instance, although OA rarely occurs in youth, people with sports injuries younger than 30 years old are at increased risk [18].
2.2. Obesity
Individuals living with obesity have a 66% chance of developing symptomatic knee OA compared to a 45% chance of developing OA for people with a conventional weight. In addition, the Framingham OA study [19] shows that women who lost about 5 kg—2 units of body mass index—reduced their risk of knee OA by a half [18]. Obesity increases the risk of developing OA through systemic and biomechanical factors. For instance, obesity alters (i) metabolism and joint inflammation that contributes to OA in non-weight-bearing joints such as the hands and (ii) biomechanical loading on weight-bearing joints such as the knee or the hip. Regarding the biomechanical factors-based on the multiplier effect of lever arms outside the body’s central axis, a force of three to six times the body weight is exerted across the knee during a single-leg stance in walking. Thus, in an individual living with obesity, the increase in weight multiplies the force across the knee during walking, putting the joint’s tissue at high risk of damage [20]. However, whether the weight has a limited effect on the progression of knee OA to moderately misaligned knees (2–7 degrees), knees that were severely misaligned would lead to an OA joint regardless of the weight added to it [21]. In addition, the correlation between obese patients and OA is further strengthened by the development of adipose tissue that secretes adipokines. Indeed, this biologically active substance contributes to joint inflammation that alters cartilage homeostasis, making them more susceptible to OA [21].
2.3. Biomechanical Load
Biomechanical overload of a joint through activities requiring repetitive and excessive joint loading, such as knee bending, is associated with knee OA. Indeed, cartilage loss is a mechanically mediated process that is more likely to occur in areas of high stress [8], where an increased expression of cytokines, chemokines, and proteolytic enzymes—PICs and MMPs—was found in response to high fluid shear stress [22]. Similarly, an increase in pressure on the posterior horn of the meniscus during occupational activities with deep flexion loading initiates the degenerative process in the joint [23]. For instance, high-impact sports activities, such as hockey, football, and soccer, lead to undue stress on joints and increase knee OA risk in adults [17][24]. While deep squatting has been shown to increase compressive and posterior shear forces on the knee, 7 and 5 times the body weight, respectively, it is not yet proven that it leads to OA [25].
Joint malalignment—changes in joint geometry—decreases the joint’s ability to adapt to its biomechanical environment, contributing to cartilage or bone tissue damage. For instance, varus knee malalignment and dynamic knee adduction moments have been found to cause medial compartment knee OA due to the increase in mechanical stress on the medial compartment of the knee; the reverse is valid for a valgus knee alignment [26][27][28]. In addition, leg length discrepancies lead to asymmetrical joint mechanics during weight-bearing activities, contributing to the development of hip OA. To compensate for the differences, an individual may increase knee flexion or hip adduction of the longer limb during stance, increasing the force at those joints [17][25].
The biomechanical load can also lead to sports-related joint injuries, a risk factor for OA. For instance, the lack of a functionally standard ACL or meniscus changes the static and dynamic loading of the knee, generating increased forces on the cartilage and SB, leading to OA [29]. Indeed, among Swedish soccer players, radiographic OA—14 years after injuring the ACL—was present in 41% of injured knees compared to 4% in uninjured knees (no difference if there was surgical intervention). In relation to this, in long-term follow-up studies of young athletes with meniscus surgery, more than 50% had OA and associated pain and functional impairment [25]. Thus, OA is increasingly thought of as joint failure driven by abnormal joint loading rather than a discrete disease entity. It is more and more considered a primarily mechanical problem, where the risk factors are all found to affect the biomechanical loading of the joint, contributing to the disease progression.
References
- Buckwalter, J.A.; Mankin, H.J.; Grodzinsky, A.J. Articular cartilage and Osteoarthritis. Instr. Course Lect. 2005, 54, 465–480.
- Brandt, K.D.; Dieppe, P.; Radin, E. Etiopathogenesis of Osteoarthritis. Med. Clin. N. Am. 2009, 93, 1–24.
- Wieland, H.A.; Michaelis, M.; Kirschbaum, B.J.; Rudolphi, K.A. Osteoarthritis—An untreatable disease? Nat. Rev. Drug Discov. 2005, 4, 331–344.
- Poulet, B.; Hamilton, R.W.; Shefelbine, S.; Pitsillides, A.A. Characterizing a novel and adjustable noninvasive murine joint loading model. Arthritis Rheum. 2011, 63, 137–147.
- Yu, H.; Huang, T.; Lu, W.W.; Tong, L.; Chen, D. Osteoarthritis Pain. IJMS 2022, 23, 4642.
- Zhang, Y.; Nevitt, M.; Niu, J.; Lewis, C.; Torner, J.; Guermazi, A.; Roemer, F.; McCulloch, C.; Felson, D. Fluctuation of knee pain and changes in bone marrow lesions, effusions, and synovitis on magnetic resonance imaging. Arthritis Rheum. 2011, 63, 691–699.
- Adatia, A.; Rainsford, K.D.; Kean, W.F. Osteoarthritis of the knee and hip. Part I: Aetiology and pathogenesis as a basis for pharmacotherapy. J. Pharm. Pharmacol. 2012, 64, 617–625.
- Neogi, T. Clinical significance of bone changes in Osteoarthritis. Ther. Adv. Musculoskelet. 2012, 4, 259–267.
- Li, G.; Yin, J.; Gao, J.; Cheng, T.S.; Pavlos, N.J.; Zhang, C.; Zheng, M.H. Subchondral bone in Osteoarthritis: Insight into risk factors and microstructural changes. Arthritis Res. Ther. 2013, 15, 223.
- Hargrave-Thomas, E.; van Sloun, F.; Dickinson, M.; Broom, N.; Thambyah, A. Multi-scalar mechanical testing of the calcified cartilage and subchondral bone comparing healthy vs early degenerative states. Osteoarthr. Cartil. 2015, 23, 1755–1762.
- Botter, S.; Glasson, S.; Hopkins, B.; Clockaerts, S.; Weinans, H.; van Leeuwen, J.; van Osch, G. ADAMTS5−/− mice have less subchondral bone changes after induction of Osteoarthritis through surgical instability: Implications for a link between cartilage and subchondral bone changes. Osteoarthr. Cartil. 2009, 17, 636–645.
- Fang, H.; Huang, L.; Welch, I.; Norley, C.; Holdsworth, D.W.; Beier, F.; Cai, D. Early Changes of Articular Cartilage and Subchondral Bone in The DMM Mouse Model of Osteoarthritis. Sci. Rep. 2018, 8, 2855.
- Amini, M.; Nazemi, S.M.; Lanovaz, J.L.; Kontulainen, S.; Masri, B.A.; Wilson, D.R.; Szyszkowski, W.; Johnston, J.D. Individual and combined effects of OA-related subchondral bone alterations on proximal tibial surface stiffness: A parametric finite element modeling study. Med. Eng. Phys. 2015, 37, 783–791.
- Castañeda, S.; Roman-Blas, J.A.; Largo, R.; Herrero-Beaumont, G. Subchondral bone as a key target for osteoarthritis treatment. Biochem. Pharmacol. 2012, 83, 315–323.
- Goldring, M.; Goldring, S.R. Articular cartilage and subchondral bone in the pathogenesis of Osteoarthritis: Articular cartilage and subchondral bone. Ann. N. Y. Acad. Sci. 2010, 1192, 230–237.
- Cisternas, M.G.; Murphy, L.; Sacks, J.J.; Solomon, D.H.; Pasta, D.J.; Helmick, C.G. Alternative Methods for Defining Osteoarthritis and the Impact on Estimating Prevalence in a US Population-Based Survey: OA Prevalence in a Population-Based Survey. Arthritis Care Res. 2016, 68, 574–580.
- Caine, D.J.; Golightly, Y. Osteoarthritis as an outcome of paediatric sport: An epidemiological perspective. Br. J. Sports Med. 2011, 45, 298–303.
- Murphy, L.; Helmick, C.G. The Impact of Osteoarthritis in the United States: A Population-Health Perspective A population-based review of the fourth most common cause of hospitalization in US adults. Orthop. Nurs. 2012, 31, 85–91.
- Felson, D.T. The epidemiology of knee osteoarthritis: Results from the framingham osteoarthritis study. Semin. Arthritis Rheum. 1990, 20, 42–50.
- Berenbaum, F.; Wallace, I.J.; Lieberman, D.E.; Felson, D.T. Modern-day environmental factors in the pathogenesis of Osteoarthritis. Nat. Rev. Rheumatol. 2018, 14, 674–681.
- Felson, D.T.; Goggins, J.; Niu, J.; Zhang, Y.; Hunter, D.J. The effect of body weight on progression of knee osteoarthritis is dependent on alignment. Arthritis Rheum. 2004, 50, 3904–3909.
- Wang, P.; Guan, P.; Guo, C.; Zhu, F.; Konstantopoulos, K.; Wang, Z. Fluid shear stress-induced osteoarthritis: Roles of cyclooxygenase-2 and its metabolic products in inducing the expression of proinflammatory cytokines and matrix metalloproteinases. FASEB J. 2013, 27, 4664–4677.
- Ezzat, A.M.; Li, L.C. Occupational Physical Loading Tasks and Knee Osteoarthritis: A Review of the Evidence. Physiother. Can. 2014, 66, 91–107.
- Caine, D.; Meyers, R.; Nguyen, J.; Schöffl, V.; Maffulli, N. Primary Periphyseal Stress Injuries in Young Athletes: A Systematic Review. Sports Med. 2022, 52, 741–772.
- Nagura, T.; Matsumoto, H.; Kiriyama, Y.; Chaudhari, A.; Andriacchi, T.P. Tibiofemoral Joint Contact Force in Deep Knee Flexion and Its Consideration in Knee Osteoarthritis and Joint Replacement. J. Appl. Biomech. 2006, 22, 305–313.
- Miyazaki, T. Dynamic load at baseline can predict radiographic disease progression in medial compartment knee osteoarthritis. Ann. Rheum. Dis. 2002, 61, 617–622.
- Sharma, L. Osteoarthritis of the Knee. N. Engl. J. Med. 2021, 384, 51–59.
- Sharma, L.; Song, J.; Felson, D.T.; Cahue, S.; Shamiyeh, E.; Dunlop, D.D. The role of knee alignment in disease progression and functional decline in knee osteoarthritis. JAMA 2001, 286, 188–195.
- Lohmander, L.S.; Englund, P.M.; Dahl, L.L.; Roos, E.M. The Long-term Consequence of Anterior Cruciate Ligament and Meniscus Injuries: Osteoarthritis. Am. J. Sports Med. 2007, 35, 1756–1769.
More
Information
Subjects:
Ophthalmology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
690
Revision:
1 time
(View History)
Update Date:
13 Jun 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No