Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Javier Martínez-García | -- | 1168 | 2022-06-09 20:17:09 | | | |
| 2 | Catherine Yang | Meta information modification | 1168 | 2022-06-10 03:16:00 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Martínez-García, J.; , .; Smerdou, C. Cholestatic Diseases. Encyclopedia. Available online: https://encyclopedia.pub/entry/23900 (accessed on 24 July 2026).
Martínez-García J, , Smerdou C. Cholestatic Diseases. Encyclopedia. Available at: https://encyclopedia.pub/entry/23900. Accessed July 24, 2026.
Martínez-García, Javier, , Cristian Smerdou. "Cholestatic Diseases" Encyclopedia, https://encyclopedia.pub/entry/23900 (accessed July 24, 2026).
Martínez-García, J., , ., & Smerdou, C. (2022, June 09). Cholestatic Diseases. In Encyclopedia. https://encyclopedia.pub/entry/23900
Martínez-García, Javier, et al. "Cholestatic Diseases." Encyclopedia. Web. 09 June, 2022.
Copy Citation
Cholestatic diseases are based on bile dysfunction due to defects affecting bile synthesis or secretion. These processes involve a wide range of enzymes and membrane transporters involved in hepatobiliary circulation. According to its origin, cholestasis can be classified into two main groups: acquired cholestasis and genetic cholestasis.
cholestatic diseases
therapy
1. Acquired Cholestasis
Most cholestatic diseases are acquired, presenting a dysregulation of the hepatobiliary transporters as a consequence of an adaptive and protective response to bile acid (BA) accumulation in the liver. This regulation is multifactorial, involving different elements such as hormones, BAs, proinflammatory cytokines, and drugs. These different factors mediate the activation of transcription factors that regulate the expression of export pumps, which promote the reduction of intracellular BAs by their excretion in the urine, resulting in the detoxification of the liver [1]. Acquired cholestatic diseases include primary biliary cholangitis (PBC), primary sclerosing cholangitis (PSC), intrahepatic cholestasis of pregnancy (ICP), biliary atresia, drug-induced cholestasis, and inflammation-mediated cholestasis [2][3].
PBC and PSC are classified as autoimmune diseases of the hepatobiliary system, characterized by the presence of antimitochondrial antibodies, portal inflammation, and an immune-mediated destruction of intra- and extra-hepatic bile ducts [4][5]. Clinical manifestations vary widely, from asymptomatic to end-stage biliary cirrhosis. The pathogenesis of the disease is multifactorial, involving genetic, epigenetic, and environmental factors [4][6].
ICP, which is the most common disorder of the hepatobiliary system, is characterized by high serum BA levels in the third trimester of pregnancy that cause severe pruritus. In the development of this cholestatic disorder, high levels of gestational hormones, such as estrogen and progesterone, play a major causative role, while genetic factors may also be involved. Although symptoms disappear after childbirth, the biliary disorder can often recur during future pregnancies [7].
Biliary atresia is a rare liver disease affecting the bile ducts, resulting in the main cause of neonatal cholestasis. The etiology of this biliary disorder is unknown. In some cases, the origin is thought to be due to an exacerbated autoimmune response in the bile duct epithelium as a consequence of a viral infection or due to toxin-induced injury after birth [8]. In other cases, it is thought to be due to a malformation of the bile ducts during gestation. However, it is known that an early diagnosis allows for better outcomes after surgery [9].
Finally, drug- and inflammation-induced cholestasis are closely related. Both drugs and proinflammatory agents can induce cholestasis following inhibition of hepatobiliary transporters but rarely result in severe liver injury. These types of cholestasis have an immunological origin mediated by proinflammatory cytokines directed against the bile duct epithelium that can alter BA secretion [10].
2. Inherited Cholestasis
Genetic cholestasis, which represents a minority of all cholestatic disorders, includes different types of progressive familial intrahepatic cholestasis (PFIC) associated with mutations in relevant channel transporters of the hepatobiliary system. PFIC is a heterogeneous group of autosomal recessively inherited monogenic disorders with a low incidence of 1:50,000–100,000 births worldwide, representing approximately 15% of all cases of neonatal cholestasis [11]. These cholestatic syndromes are characterized by an early onset of the disease, usually in infancy, associated with clinical manifestations such as pruritus, jaundice, malabsorption of fat and fat-soluble vitamins, and hepatomegaly [11]. PFIC is associated with several liver complications, such as portal hypertension and cirrhosis, and can progress to end-stage liver disease and liver failure between childhood and adulthood. Depending on the type of PFIC, extrahepatic clinical manifestations or hepatocellular carcinoma (HCC) may occur [12]. The most common biochemical features of this group of hepatobiliary diseases are increased serum BAs and bilirubin [11]. Depending on their genetic origin, PFICs can be classified into six types. Mutations in ATP8B1, ABCB11, ABCB4, tight junction protein 2 (TJP2), NR1H4, and Myosin VB (MYO5B) genes are known to be the cause of PFIC 1-6 types, respectively (Figure 1). In PFIC1, mutations in the familial intrahepatic cholestasis 1 (FIC1) gene cause the loss of the asymmetric distribution of phospholipid content in the canalicular membrane, leading to membrane destabilization and reduced BA transport, resulting in their accumulation in hepatocytes, causing cholestasis. Mutations in the ABCB11 gene can result in PFIC2 due to the absence of a functional bile salt export pump (BSEP) protein, which also leads to toxic accumulation of BA in hepatocytes. In PFIC3, mutations in ABCB4 cause multidrug resistance protein 3 (MDR3, ABCB4) deficiency, which results in low levels of phosphatidylcholine (PC) in the bile, which is needed to form micelles and neutralize the toxicity of hydrophobic BAs, resulting in damage to the epithelium of bile canaliculi. Mutations in TJP2 lead to the misdistribution of claudin tight junction in canaliculi, resulting in bile leakage and subsequently in PFIC4. PFIC5 is due to mutations in the NR1H4 gene that cause deficiency in farnesoid X receptor (FXR), resulting in a reduction of BSEP and ABCB4 expression and the accumulation of toxic BAs in the hepatocytes. Finally, mutations in MYO5B interfere with the processing of normal intracellular trafficking of BSEP, reducing its expression and activity at the canalicular membrane, which results in the accumulation of toxic BAs in hepatocytes, giving rise to PFIC6 [13]. Different disease characteristics such as the age of onset, severity, and the manifestation of specific complications and serum markers vary between PFIC types [12][13].
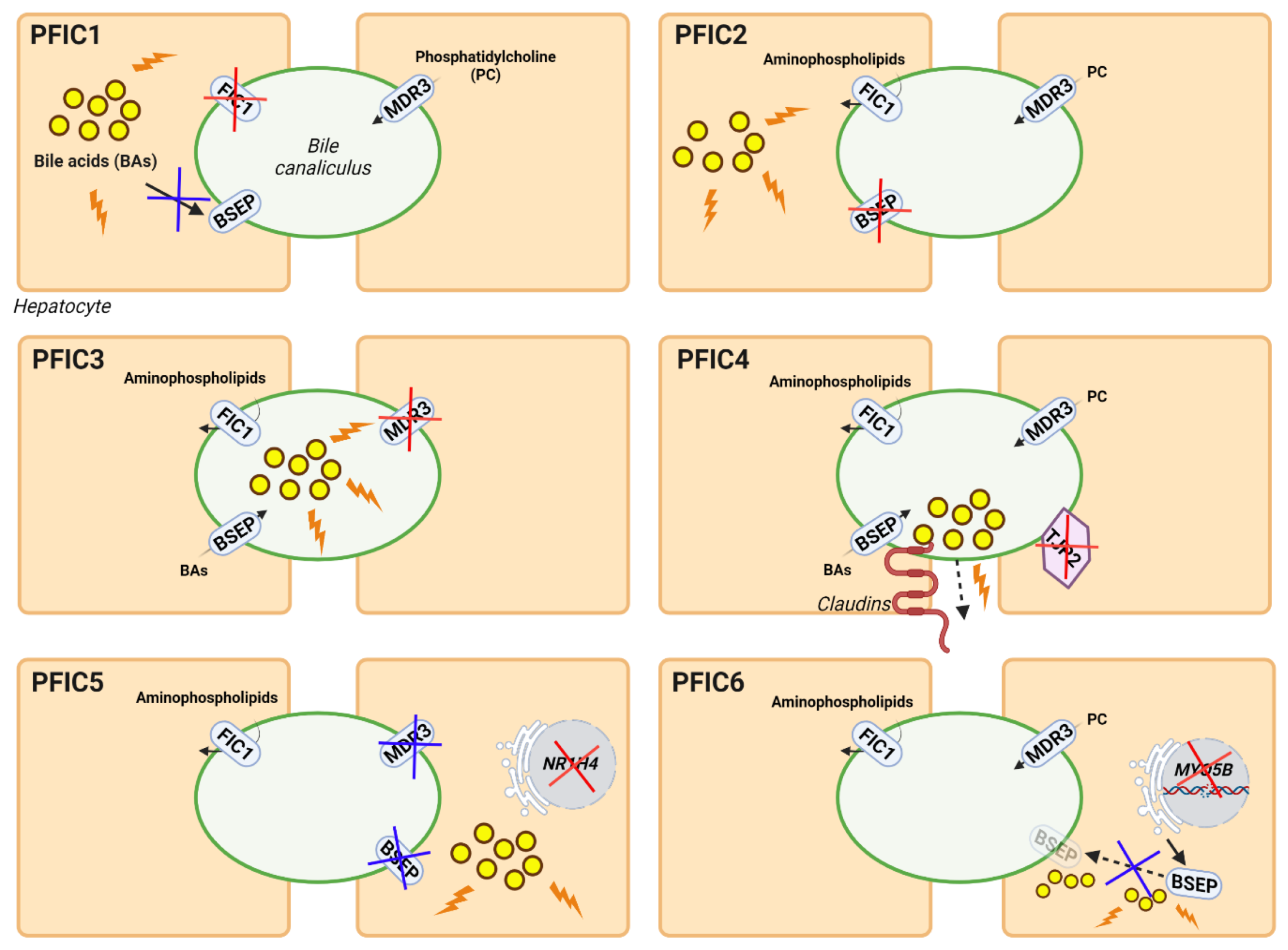
Figure 1. Genetic classification and pathogenesis of PFIC. The diagrams show the genes and functions altered in each type of PFIC. The main deficient proteins for each type of PFIC are indicated by red crosses, while derived alterations in other proteins or pathways are indicated by blue crosses. Damage due to the abnormal accumulation of BAs is shown as yellow circles with orange lightnings.
The role of BSEP in the functioning of the hepatobiliary system is very important, as mutations in different genes involved in BA metabolism and transport, such as ABCB11, NR1H4, and MYO5B causing its deficiency, cause PFIC [14][15][16]. In addition, depending on the severity of the disease, inherited intrahepatic cholestasis resulting from mutations in ATP8B1 or ABCB11 can be classified as either PFIC1 or 2, respectively, or benign recurrent intrahepatic cholestasis (BRIC) 1 or 2, respectively. Sometimes it is clinically difficult to discern between PFIC and BRIC because, in both cases, patients may present mild cholestasis with long-term complications [17]. In addition, some missense mutations in less conserved regions of the ABCB11 and ABCB4 genes promote the development of more moderate variants of cholestasis such as BRIC2, ICP, cholesterol cholelithiasis, drug-induced cholestasis, adult biliary cirrhosis, transient neonatal cholestasis, and others [18][19]. In addition, mutations in cholangiocyte transporter genes (e.g., the cystic fibrosis transmembrane conductance regulator (CFTR) gene) can cause cholestasis. In fact, a direct association between cystic fibrosis and cholestatic conditions, such as bile duct complications, gallstones, and primary sclerosing cholangitis, has been observed due to mutations in CFTR [20]. Other genetic multisystemic diseases associated with cholestatic disorders include Alagille syndrome (ALGS) and cerebrotendinous xanthomatosis (CTX). ALGS arises due to mutations in genes involved in the Notch signaling pathway, such as JAG1 and NOTCH2, and the majority of patients present cholestasis and a deficiency of bile ducts [21]. CTX is caused by mutations in the CYP27A1 gene, resulting in impaired BA biosynthesis and the accumulation of toxic metabolites. Although liver damage is not common in all CTX patients, some cases of severe infantile cholestasis have been reported [22].
References
- Lee, J.; Boyer, J.L. Molecular Alterations in Hepatocyte Transport Mechanisms in Acquired Cholestatic Liver Disorders. Semin. Liver Dis. 2000, 20, 373–384.
- Zollner, G.; Trauner, M. Mechanisms of Cholestasis. Clin. Liver Dis. 2008, 12, 1–26.
- Yokoda, R.T.; Rodriguez, E.A. Review: Pathogenesis of Cholestatic Liver Diseases. World J. Hepatol. 2020, 12, 423–435.
- Kaplan, M.M.; Gershwin, M.E. Primary Biliary Cirrhosis. N. Engl. J. Med. 2005, 353, 1261–1273.
- Gulamhusein, A.F.; Hirschfield, G.M. Primary Biliary Cholangitis: Pathogenesis and Therapeutic Opportunities. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 93–110.
- Dyson, J.K.; Beuers, U.; Jones, D.E.J.; Lohse, A.W.; Hudson, M. Primary Sclerosing Cholangitis. Lancet 2018, 391, 2547–2559.
- Reyes, H.; Sjövall, J. Bile Acids and Progesterone Metabolites Intrahepatic Cholestasis of Pregnancy. Ann. Med. 2000, 32, 94–106.
- Pang, S.-Y.; Dai, Y.-M.; Zhang, R.-Z.; Chen, Y.-H.; Peng, X.-F.; Fu, J.; Chen, Z.-R.; Liu, Y.-F.; Yang, L.-Y.; Wen, Z.; et al. Autoimmune Liver Disease-Related Autoantibodies in Patients with Biliary Atresia. World J. Gastroenterol. 2018, 24, 387–396.
- Abbey, P.; Kandasamy, D.; Naranje, P. Neonatal Jaundice. Indian J. Pediatr. 2019, 86, 830–841.
- Visentin, M.; Lenggenhager, D.; Gai, Z.; Kullak-Ublick, G.A. Drug-Induced Bile Duct Injury. Biochim. Biophys. Acta-Mol. Basis Dis. 2018, 1864, 1498–1506.
- Jacquemin, E. Progressive Familial Intrahepatic Cholestasis. Clin. Res. Hepatol. Gastroenterol. 2012, 36, S26–S35.
- Srivastava, A. Progressive Familial Intrahepatic Cholestasis. J. Clin. Exp. Hepatol. 2014, 4, 25–36.
- Amirneni, S.; Haep, N.; Gad, M.A.; Soto-Gutierrez, A.; Squires, J.E.; Florentino, R.M. Molecular Overview of Progressive Familial Intrahepatic Cholestasis. World J. Gastroenterol. 2020, 26, 7470–7484.
- Imagawa, K.; Hayashi, H.; Sabu, Y.; Tanikawa, K.; Fujishiro, J.; Kajikawa, D.; Wada, H.; Kudo, T.; Kage, M.; Kusuhara, H.; et al. Clinical Phenotype and Molecular Analysis of a Homozygous ABCB11 Mutation Responsible for Progressive Infantile Cholestasis. J. Hum. Genet. 2018, 63, 569–577.
- Gomez-Ospina, N.; Potter, C.J.; Xiao, R.; Manickam, K.; Kim, M.-S.; Kim, K.H.; Shneider, B.L.; Picarsic, J.L.; Jacobson, T.A.; Zhang, J.; et al. Mutations in the Nuclear Bile Acid Receptor FXR Cause Progressive Familial Intrahepatic Cholestasis. Nat. Commun. 2016, 7, 10713.
- Gonzales, E.; Taylor, S.A.; Davit-Spraul, A.; Thébaut, A.; Thomassin, N.; Guettier, C.; Whitington, P.F.; Jacquemin, E. MYO5B Mutations Cause Cholestasis with Normal Serum Gamma-glutamyl Transferase Activity in Children without Microvillous Inclusion Disease. Hepatology 2017, 65, 164–173.
- Luketic, V.A.; Shiffman, M.L. Benign Recurrent Intrahepatic Cholestasis. Clin. Liver Dis. 2004, 8, 133–149.
- Lam, P.; Soroka, C.; Boyer, J. The Bile Salt Export Pump: Clinical and Experimental Aspects of Genetic and Acquired Cholestatic Liver Disease. Semin. Liver Dis. 2010, 30, 125–133.
- Sticova, E.; Jirsa, M. ABCB4 Disease: Many Faces of One Gene Deficiency. Ann. Hepatol. 2020, 19, 126–133.
- Feranchak, A.P.; Sokol, R.J. Cholangiocyte Biology and Cystic Fibrosis Liver Disease. Semin. Liver Dis. 2001, 21, 471–488.
- Mitchell, E.; Gilbert, M.; Loomes, K.M. Alagille Syndrome. Clin. Liver Dis. 2018, 22, 625–641.
- Zhang, P.; Zhao, J.; Peng, X.-M.; Qian, Y.-Y.; Zhao, X.-M.; Zhou, W.-H.; Wang, J.-S.; Wu, B.-B.; Wang, H.-J. Cholestasis as a Dominating Symptom of Patients with CYP27A1 Mutations: An Analysis of 17 Chinese Infants. J. Clin. Lipidol. 2021, 15, 116–123.
More
Information
Subjects:
Gastroenterology & Hepatology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Revisions:
2 times
(View History)
Update Date:
10 Jun 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No