 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Suresh Ramakrishna | + 8393 word(s) | 8393 | 2020-10-05 02:45:55 | | | |
| 2 | Bruce Ren | + 1 word(s) | 8394 | 2020-10-09 09:49:09 | | |
Video Upload Options
Since the discovery of the ubiquitin proteasome system (UPS), the roles of ubiquitinating and deubiquitinating enzymes (DUBs) have been widely elucidated. The ubiquitination of proteins regulates many aspects of cellular functions such as protein degradation and localization, and also modifies protein-protein interactions. DUBs cleave the attached ubiquitin moieties from substrates and thereby reverse the process of ubiquitination. The dysregulation of these two paramount pathways has been implicated in numerous diseases, including cancer. Attempts are being made to identify inhibitors of ubiquitin E3 ligases and DUBs that potentially have clinical implications in cancer, making them an important target in the pharmaceutical industry. Therefore, studies in medicine are currently focused on the pharmacological disruption of DUB activity as a rationale to specifically target cancer-causing protein aberrations. Here, we briefly discuss the pathophysiological and physiological roles of DUBs in key cancer-related pathways. We also discuss the clinical applications of promising DUB inhibitors that may contribute to the development of DUBs as key therapeutic targets in the future.
1. Introduction
Cancer is fundamentally a genetic disorder that has been well characterized over the past decade. The genetic basis of different cancers has been investigated thoroughly with the application of high-throughput genome-wide association studies (GWAS), which have identified over 450 genetic variations associated with cancer risk [1]. These mutations cause cancer susceptibility, not cancer per se, providing a “head start” to the neoplastic process [2]. Germline or somatic mutations in three major classes of genes, oncogenes, tumor suppressor genes, and DNA stability genes, initiate the neoplastic process, resulting in rounds of clonal expansion and subsequent somatic mutations associated with tumor formation [3].
Proto-oncogenes are required for normal cellular homeostasis and functions. Mutations in proto-oncogenes resulting from chromosomal translocations or gene fusions spark constitutive expression under conditions where wild-type genes would be inactive [4][5]. Cancer cells, therefore, gain competitive advantages over their non-neoplastic counterparts, while mutations in tumor suppressor genes confer neoplastic cells with competitive advantages in an opposite way. Tumor suppressor genes function to control tumor formation by the stimulation of cell death and restrain inappropriate cell division. The inactivation of tumor suppressor genes arises from missense mutations or indels at active site residues, resulting in truncated proteins or epigenetic silencing [6]. A subset of tumor suppressor genes, called caretaker genes or DNA stability genes, maintain genetic integrity through nucleotide-excision repair, mismatch repair, base-excision repair, chromosomal segregation, and mitotic recombination [7][8]. Because they encode molecules that stabilize the genome, mutations in caretaker genes can contribute to the neoplastic process. Although the accumulation of several gene mutations contributes to fully fledged cancer, a few targeted pathways are involved. Examples of these, such as receptor tyrosine kinase (RTKs) signaling and the p53 pathway, will be discussed in this review [9][10].
Other important regulators of cancer progression are cellular proteases, which play indispensable roles in various biological and pathological processes [11][12]. Proteases are highly specific enzymes that selectively carry out proteolytic processing, targeting a wide range of substrates, thereby regulating processes essential for cell survival [13]. A deficiency of proteases or misdirected spatio-temporal expression patterns can cause diverse pathologies, such as neurodegenerative diseases, arthritis, cardiovascular diseases, and cancers, making them an important focus as drug targets or diagnostic markers [14]. The entire repertoire of cellular proteases, called the “degradome,” comprise about 588 proteases divided among five catalytic classes known as the aspartyl, cysteine, metallo-, serine, and threonine proteases [15]. Most of these proteolytic enzymes catalyze the hydrolysis of peptides by targeting the α-peptide bonds between naturally occurring amino acids, while others undertake slightly different reactions. Among them lie a growing group of peptidases known as deubiquitinating enzymes (DUBs) that hydrolyze isopeptide bonds in ubiquitin and ubiquitin-like protein conjugates and, thus, have emerged as crucial regulators of ubiquitin-mediated signaling pathways and are potential drug targets in various diseases, including cancer [16][17][18].
2. Ubiquitin Proteasomal Pathways and DUBs
Ubiquitination is a very important post-translational modification that plays multifaceted roles in cancer-related pathways. The ubiquitination process is also involved in protein metabolism, apoptosis, cell-cycle progression, and transcription. It involves the covalent attachment of a 76-amino acid protein called ubiquitin (Ub) to the ε-amino groups of lysine residues in the target proteins. This conjugation reaction is catalyzed by the successive action of three enzymes: E1 or Ub-activating enzyme, E2 or Ub-conjugating enzyme, and E3 or Ub-ligase enzyme [19]. The first step of this process is the ATP-dependent activation of the Ub molecule by E1, forming a thioester bond with the C-terminal of Ub. The activated Ub is then transferred to E2, forming a thioester-linked E2-Ub intermediate, which is then positioned and transferred to the target substrate protein with the help of E3. Ub can be conjugated as monomers or multiple mono-ubiquitin adducts to either the same or different residues of the target protein that directs their fate. Lysine 48 (K48), along with K11- and K29-linked poly-ubiquitin chains, targets protein destruction via the 26S proteasome. However, K63-linked poly-ubiquitination and multiple mono-ubiquitin conjugations are mainly involved in proteasome-independent events such as DNA repair and signal transduction [20][21][22], although their involvement in proteasomal targeting has also been reported [23].
Interestingly, protein ubiquitination is a dynamic and reversible process. DUBs are responsible for the removal of ubiquitin from their target proteins, rescuing them from the degradative pathway. They are also involved in the editing, maturation, and recycling of the ubiquitin molecule post degradation [24]. Some DUBs can edit the Ub signal harbored by target proteins by trimming Ub chains. For example, A20 causes K48-linked proteasomal degradation instead of K63-linked polyubiquitination of receptor-interacting serine-threonine kinase 1 (RIPK1) [25]. DUBs are highly versatile and capable of preventing lysosomal as well as proteasomal degradative pathways, thus enhancing protein stability. The deubiquitination process maintains the pool of free mono-Ub that is recycled for the ubiquitination of misfolded proteins and is therefore important in maintaining the normal rates of proteolysis within cells. Moreover, DUBs prevent the attachment of inhibitory ubiquitin chains that compete with ubiquitinated protein substrates at the binding sites of the 26S proteasome. In a nutshell, DUBs maintain the stability of protein substrates and regulate various cellular processes, such as epistasis, gene expression, chromosome segregation, DNA repair, spermatogenesis, signaling events, and cell cycle regulation [26].
More than 100 functional DUBs have been identified across the human genome and are categorized into eight families: ubiquitin-specific proteases (USPs), ubiquitin C-terminal hydrolases (UCHs), ovarian tumor proteases (OTUs), Jab1/MPN domain-associated metallopeptidase (JAMM) domain proteins, Josephin or Machado–Joseph disease protein domain proteases (MJDs), the monocyte chemotactic protein-induced protein (MCPIP) family, the motif interacting with Ub-containing novel DUB family (MINDY), and Zn-finger and UFSP domain proteins (ZUFSPs) [27][28][29]. Figure 1 presents the phylogeny, role in cancer and other diseases, and inhibitors of fourteen USPs, the largest subfamily of DUBs.
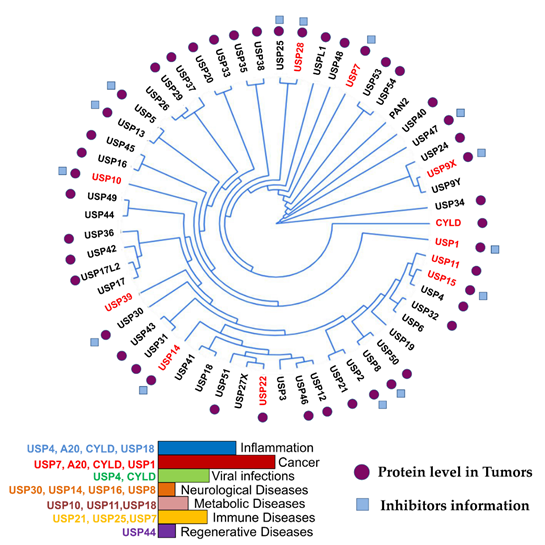
Figure 1. USP phylogenetic tree. Ubiquitin-specific proteases (USPs) and their association with different diseases are indicated in the histogram. The branches arising from one node represent a clade. The distance between the USPs in the phylogenetic tree describes the relationship among the individuals in this family. DUBs with high expression in different cancers are highlighted in red in the phylogenetic tree. Tumors expressing medium levels of USPs in different cancer types are indicated with purple circles. Blue boxes indicate USPs with identified drugs. This figure is a representation of data obtained from at least 1% of cancer patients [30].
DUBs control several aspects of cell physiology, and defects in these processes have many clinical implications. Examples of these deregulations include the BRCA1-associated protein 1 (BAP1) of the UCH family of DUBs, which is commonly mutated in mesotheliomas, melanomas, and renal cell carcinomas [31]; USP6 translocation in aneurysmal bone cysts [32]; USP9X mutations that cause developmental disorders and deregulated expression in cancer [33][34]; USP15 overexpression in certain glioblastomas, ovarian cancer, and breast cancer [35]; and cylindromatosis (CYLD) mutations in familial cylindromatosis [36]. Due to the involvement of the components of ubiquitin machinery in different cancer types, inhibitors targeting DUBs are attracting much attention from pharmaceutical industries, and several candidates have already been identified as potentially rewarding drug targets. This review focuses on the production of chemical libraries consisting of inhibitors that specifically target DUBs involved in cancer.
3. DUBs in Cancer
The functions of DUBs are not just limited to the reversal of ubiquitination but encompass multi-dimensional aspects like protein trafficking, apoptosis, chromatin remodeling, DNA damage repair, cell cycle regulation, and signaling pathway modulations that are frequently linked to the development of neoplastic diseases [37]. DUBs have been implicated in tumorigenesis at multiple levels, and, after the clinical success of the broadly acting proteasome inhibitor bortezomib, used to treat refractory multiple myelomas (MM) and myeloid cell leukemia (MCL), DUBs have become attractive targets for the development of targeted novel therapies in cancer [38][39]. DUB modifications and their implications in different cancers are described briefly here to gain insight into their role as potential drug targets.
3.1. Genetically Altered DUBs in Cancer
Genetically altered DUBs have been identified in cancers, corroborating their roles as true oncogenes and tumor suppressors. Several studies implicate USP4, USP7, USP6, USP15, USP16, USP42, and USP28 as oncogenes, whereas CYLD, A20, and BAP1 act as tumor suppressors [40][41][42]. There are reports that DUBs such as USP7, CYLD, and A20 play dual roles in cancer, acting as both anti- and pro-tumorigenic enzymes according to the affected targets [43][44][45]. USP6, or Tre2, acts as an oncogene due to chromosomal rearrangements of the osteoblast cadherin11 gene (CDH11) promoter, which cause an overexpression of USP6 linked to neoplastic aneurismal bone cysts [46]. USP6 also regulates expression of the proto-oncogene c-Jun and promotes cell invasion [47]. Somatic mutations in the USP28 gene have been identified in lobular breast cancer cases [48].
CYLD mutations have been described in familial cylindromatosis, familial trichoepithelioma, and Brooke–Spiegler syndrome, in which patients have a predisposition to develop multiple skin tumors of the neck and head [49]. Nuclear factor (NF)-κB signaling has well-established roles in cancer promotion and CYLD has been reported to negatively regulate this pathway through its deubiquitinating activity [50][51][52][53]. Several lymphoma subtypes show chromosomal deletions and inactivating mutations in the A20 gene loci. Inactivating A20 mutations have been reported in marginal zone lymphomas [54] and A20 mRNA expression loss has been found in non-Hodgkin’s lymphoma, Burkitt’s lymphoma, and T-cell lymphoma [55]. A20, also called tumor necrosis factor alpha-induced protein 3 (TNFAIP3), is a negative NF-κB signaling regulator [56]. Innate immune signals, including the tumor necrosis factors and toll-like receptors, trigger A20-mediated termination of NF-κB signaling.
BAP1, or BRCA1-associated protein, is a nuclear-localized DUB and may be the most commonly mutated DUB in cancers. Originally identified as an interacting partner of the BRCA1 tumor suppressor, BAP1 augments tumor suppressor functions but has not been identified as a DUB for BRCA1 [57]. Germline mutations in BAP1 are associated with tumor predisposition syndrome for mesotheliomas and melanocytic tumors [58][59], whereas deletions and point mutations have been identified in lung and breast cancers [60][61]. BAP1-inactivating mutations are also identified in metastasizing uveal melanomas and malignant pleural mesotheliomas [62]. BAP1 also controls cell cycle progression by the regulation of host cell factor-1 (HCF-1) and other G1-S-transition-related genes [63].
3.2. DUBs Regulating Signaling Pathways in Cancer
Signaling pathways are receiving more attention than the individual genes that are altered in cancers [64]. DUBs have a profound influence over commonly mutated cancer pathways such as p53, NF-κB, RTKs, Wnt, and transforming growth factor (TGF)-β, which are briefly described below.
3.2.1. p53 Signaling
The tumor suppressor p53 is lost or mutated across most tumor types [65]. p53 has evolved an exquisite regulatory mechanism enabling rapid stress responses and preventing errant activation, mainly due to the pivotal role that it plays in cellular activities [66]. The p53 inhibitors ARF-BP1, MDM2, Pirh2, COP1, and MDM4 of the E3 ubiquitin ligase family are central to this regulation [67]. Several DUBs have been identified as important p53 regulators. Thus far, USP2a, USP4, USP5, USP7, USP9X, USP10, USP11, USP15, USP24, USP29, and USP49 have been associated with p53 regulation [68]. USP2 and its isoforms have previously been shown to have oncogenic properties and have been extensively investigated by cancer biologists. USP2a has high expression levels in human prostate adenocarcinomas, and treatment with chemotherapeutic agents prevents the apoptosis of cancerous cells [69][70][71]. USP2 has been reported to deubiquitinate proteins involved in different pathways, such as MDM2, MDMX, apoptosis inducing factor (AIF), Cyclin D, fatty acid synthase (FASN), Aurora-A kinase, and epidermal growth factor receptor (EGFR) [72][73][74][75][76][77][78]. USP2a does not directly deubiquitinate p53 but acts on MDM2 without reducing p53 ubiquitination. MDM2 is an E3 ligase for p53 and promotes its ubiquitination and degradation; therefore, suppression of USP2a leads to the activation of p53 in vivo. However, in glioma cells, USP2a binds to MDM4, enhancing the localization of p53 to the mitochondria and promoting apoptosis [79]. Stabilization of ARF-BP1 via USP4 deubiquitination has been reported to reduce p53 levels. Enhanced apoptosis in thymus and spleen has been observed in USP4 knockout mice in response to ionizing radiation. USP4-/- mouse epithelial fibroblasts (MEFs) exhibit premature cellular senescence, retarded growth, resistance to oncogenic transformation, and hyperactive DNA damage checkpoints compared to wild-type MEFs. These observations indicate higher p53 levels and activity due to the absence of USP4. USP4 has been suggested as a potential oncogene and is observed to be highly upregulated in several cancers.
USP5 negatively regulates the level and transcriptional activity of p53. USP5 disassembles unanchored poly-ubiquitin chains from its substrate to recycle free mono-ubiquitin and contributes to K48-linked poly-ubiquitin disassembly [80]. Suppression of USP5 inhibits p53 proteasomal degradation without affecting MDM2, ultimately resulting in p53 activation. USP5 is, therefore, a potential p53-activating therapeutic drug target in cancer therapy [80]. USP7 dynamically regulates p53 as well as MDM2 [81]. Inhibition of USP7 triggers MDM2 degradation and p53 stabilization, ultimately leading to apoptotic pathway activation in tumor cells [82]. USP7 inhibitors can have alternative therapeutic mechanisms of action, as USP7 has several other targets including forkhead box protein O4 (FOXO4), FOXP3, phosphatase and tensin homolog (PTEN), and UBL SUMO [83][84][85][86]. USP10 is identified as a cytoplasmic DUB that regulates p53 levels and localization by reversing Mdm2-induced p53 nuclear export and degradation. It regulates the stability of p53 without interacting with MDM2, unlike USP2 and USP7, and has been shown to stabilize the wild type as well as the mutated form of p53, playing a dual role in tumorigenesis, which depends upon the status of p53 [87]. USP28 is highly expressed in many cancer types, such as in breast cancer [88], non-small-cell lung cancer (NSCLC) [89], gastric cancer [90], bladder cancer [91], and colorectal cancer [92]. USP28 is an indirect regulator of the proto-oncogene c-Myc via E3 ligase Fbw7a as well as JUN and Notch [93]. USP28 is also found to regulate lysine-specific demethylase 1 (LSD1) protein levels in gastric cancer cells and has also been connected to p53 via TP53-binding protein 1 (TP53BP1) [94]. Due to the role played by USP28 in various malignancies, it is identified as a potential drug target for cancer treatments. In response to oxidative stress, USP29 deubiquitinates and stabilizes p53 by reversing MDM2-directed p53 ubiquitination [95].
USP42 was identified as a p53 DUB by siRNA library screening and deubiquitinates p53 by removing Mdm2-conjugated Ub [96]. It stabilizes p53 and helps recruit p53-interacting factors contributing to p53 functions [97]. USP9X attenuates the degradation of p53 and controls several cellular functions, including proliferation, apoptosis, and chemo-resistance. Studies have shown that USP9X mediates apoptosis and/or survival in p53-deficient cells [98]. USP24 regulates p53 and the apoptosis pathway related to p53 function in the DNA damage response [99]. USP15 upregulates transcriptional activation and the stability of p53 in response to TGF-β signaling, thereby affecting the stability of both Mdm2 and p53 [100]. USP11 stabilizes and activates p53 following DNA damage by targeting ubiquitinated p53. Post DNA damage, USP11 activates p53 and its target genes such as Puma, Bax, and p21 [101]. USP49 has also been reported as a novel positive regulator of p53 transcriptional activity and stability. It has been shown to render HCT119 cells more sensitive to DNA damage induced by etoposide and is upregulated in response to DNA damage and other cell stresses, thus acting as a potential tumor suppressor and forming a positive feedback loop with p53 [102].
3.2.2. Nuclear Factor-κB Signaling
The NF-κB transcription factor family members have been recognized as crucial players in cancer initiation and progression while also playing roles in inflammation and innate immunity [103]. A20 and CYLD act on several components of the pathway, downregulating NF-κB signaling and thereby acting as tumor suppressors [104]. Several intermediates of the NF-κB signaling pathway, NEMO, TRAF-2, and TRAF-6, undergo K63 poly-ubiquitination post-receptor activation and activate the IκB kinase (IKK). IKK, in turn, phosphorylates NF-κB inhibitor IκB, leading to its dissociation from NF-κB, K48-linked poly-ubiquitination by the phospho-specific E3 ligase β-TrCP, and finally its proteasomal degradation [105]. CYLD potentially dampens NF-κB signaling following the removal of K63 poly-ubiquitin chains from TRAF-2, TRAF-6, and NEMO. The generation of mutant CYLD-/- mice has confirmed the role of this DUB as a tumor suppressor. The ability of CYLD to prevent cell proliferation was observed by the effect on skin tumor formation. A single 12-O-tetradecanoylphorbol-13 acetate (TPA) application resulted in an increase in Ki-67 and cyclin D1 in skin sections of CYLD-/- animals [106]. Colon tumor induction in CYLD-/- mice has been linked to inflammation associated with enhanced NF-κB activity [107]. These studies collectively strengthen the role of CYLD as an important tumor suppressor in humans.
A20 regulates NF-κB pathways through its ovarian tumor deubiquitinase (OTU) domain and zinc-finger domain [108][109]. The OTU domain is responsible for deubiquitinating NF-κB intermediates such as IKKg, RIP1, RIP2, TRAF2, and TRAF6, and shows a stronger preference for K63-linked Ub chains. The zinc-finger domain is responsible for the Ub ligase activity of A20, replacing the K63-linked chains with proteasome-targeting K48 chains [110][111]. USP21 is another DUB identified as an inhibitor of the NF-κB pathway, which interacts and deubiquitinates RIP1 in a DUB-dependent manner [112]. USP4 has an important role in the downregulation of TNFα-induced activation of NF-κB via TGF-β-activated kinase 1 deubiquitination [113]. Cezanne interacts with DJ-1 and targets RIPK1 signaling intermediates, thereby suppressing NF-κB nuclear translocation and its activity [114][115]. USP31 deubiquitinates K63-linked poly-ubiquitinated proteins of the TNF-induced NF-κB pathway [116]. USP7 deubiquitinates TRAF6 and NEMO, thus negatively regulating NF-κB in the TLR pathway while USP2 acts as a positive regulator of TNF-induced NF-κB activation[117][118]. USP2 is required for IκB phosphorylation and the nuclear translocation of NF-κB [118]. OTUD5 targets TRAF3 for deubiquitination, resulting in diminished IL-10 and type I interferon responses[119]. USP15 stabilizes IκBa, whereas USP11 interacts with the inhibitor IKKa upon TNFα induction [120[121]. MCP-induced protein 1 (MCPIP1) is a novel DUB that targets TRAF2 and TRAF6, which are essential for termination of NF-κB and JNK signaling .
3.2.3. Receptor Tyrosine Kinase Signaling
Growth factor receptors are composed mainly of transmembrane, extracellular, and cytoplasmic tyrosine kinase (TK) domains. RTK activation is required for the regulation of key processes, such as cell growth, survival, tissue repair, and regeneration; therefore, dysregulation of this pathway has been reported in numerous cancers. The significance of the RTK pathway makes it an attractive therapeutic target [122]. The mechanism responsible for the delivery of membrane receptors from the cell surface to lysosomes has been intensively studied for EGFR, and ubiquitination serves as a key signal for this pathway. The trafficking of RTKs such as Met, ErbB2, and EGFR is regulated by DUBs [123]. USP8 stabilizes RTKs by deubiquitinating EGFR on early endosomes, thus allowing their recycling to the plasma membrane [124]. USP8 has also been reported to promote RTK degradation [125]. RNA interference screens have identified USP18 as an EGFR synthesis regulator and POH1 as an ErbB2 regulator [126][127]. USP18 regulates the expression of EGFR and is responsible for cancer cell survival through mRNA stabilization and transcriptional activation of miR-7 host genes [128]. USP9X is a DUB for Eps15, which is essential for EGFR internalization, thereby acting as an EGFR regulator [129]. Cezanne-1 was identified as an EGFR-regulating DUB using RNA interference screening and is reported to be overexpressed in breast cancer and in approximately one-third of mammary tumors. In conclusion, oncogenic growth signals are promoted following deubiquitination by Cezanne-1, which curtails the degradation of growth factor receptors [130].
3.2.4. Wnt Signaling
Another frequently altered pathway in cancer is the Wnt signaling pathway, which essentially controls embryonic development [131]. Malignant transformation, tumor progression, and resistance to conventional cancer therapy are favored by dysregulation of canonical and non-canonical Wnt-planar cell polarity (PCP) signaling [132][133]. Aberrant Wnt signaling is also suggested to subvert cancer immune surveillance, promoting immune evasion and resistance to immunotherapies and immune checkpoint blockers [134][135][136]. Regulation by ubiquitination and deubiquitination play key roles in canonical Wnt signaling and in non-canonical Wnt-PCP signaling pathways. The ubiquitin-mediated system regulates cytoplasmic β-catenin as well as other proteins upstream of the Wnt pathway [137][138]. USP9X has been reported as a DUB for DVL2, which is required for the activation of canonical Wnt. Increased DVL2 ubiquitination activates the PCP pathway via localization to actin-rich projections, making USP9X an important therapeutic target for cancers [139]. A strong correlation has been found between the levels of β-catenin and USP14 by tissue microarray analysis of colon cancers, suggesting an oncogenic role of USP14 by the enhancement of Wnt/β-catenin signaling [140]. USP6 was identified by genome-wide siRNA screening as a potent activator of Wnt signaling by deubiquitinating Fzds, thereby increasing their cell surface abundance [141]. USP4 is a potential cancer therapy target as it deubiquitinates and facilitates the nuclear localization of β-catenin, acting as a positive regulator of the Wnt/β-catenin pathway [141][142]. USP34 hinders β-catenin-dependent transcription, acting as a positive Wnt signaling regulator [143].
3.2.5. Transforming Growth Factor-β Signaling
The multifunctional protein TGF-β plays a dual role in oncogenesis, acting as an anti-proliferative factor during the early stages and as an epithelial-to-mesenchymal transition (EMT)-promoting factor in the later stages [144]. So far, only a few DUBs have been reported to regulate this pathway. USP9X acts as a DUB for SMAD4, promoting its association with SMAD2 and positively regulating TGF-β [145]. USP9X also deubiquitinates NAUK1 and MARK4, AMPK-related kinases important for the regulation of cell proliferation and polarity, by modulating their phosphorylation and activation by LKB1 [146]. USP15, thought to be important for the proliferation of glioblastoma cells, deubiquitinates the type I TGFβ receptor via binding to the SMAD7-SMAD-specific E3 ubiquitin ligase 2 (SMURF2) complex. Overexpression of USP15, which correlates with higher TGF-β activity, is mostly found in ovarian cancer, breast cancer, and glioblastomas. Reducing TGF-β signaling by depleting USP15 reduces the oncogenicity of patient-derived glioma-initiating cells, indicating the therapeutic capabilities of USP15 inhibition [147]. USP15 has also been reported to deubiquitinate other TGF-β signaling components and receptor-regulated SMADs (R-SMAD), whereas UCHL5 and AMSH-LP potentiate TGF-β responses through I-SMADs interactions [147][148][149].
3.3. DUBs in Cell Cycle Regulation
The regulation of proteins such as cyclins, cyclin-dependent kinases (CDKs), and other checkpoint molecules that mediate the cell cycle is highly important to ensure proper division of cells. Inappropriate expression or deregulation of these proteins can result in different types of tumors. Several studies have reported the role of DUBs in regulating cell cycle proteins. DUBs such as CYLD, USP5, USP13, USP15, USP17, USP37, USP39, and USP44 are key regulators of events that occur during mitosis. CYLD, USP15, and USP44 modulate spindle formation. USP44 deubiquitinates anaphase-promoting complex (APC) coactivator Cdc20 and prevents chromosomal segregation errors [150]. Mutations in USP44 result in the formation of tumors, especially in lung cancer [150]. USP37 promotes entry into the S phase of the cell cycle by anaphase-promoting complex (APC) coactivator Cdh1 [151]. USP15 regulates the cell cycle by stabilizing and rescuing the expression of newly synthesized REST at mitotic exit [152]. USP17 is tightly controlled in the cell cycle and its knockdown attenuates GTPase signaling and proliferation of tumor-derived cell lines [153]. USP39 is important for the integrity of the mitotic spindle checkpoint, and depleting USP39 during mRNA processing reduces mRNA levels of Aurora B [154]. CYLD regulates polo-like kinase 1 (Plk1) and regulates mitotic entry [155]. USP3 is required for chromatin modification and S phase progression [156]. USP2 prevents ubiquitin-mediated degradation of cyclin-D1. USP2a deubiquitinates and stabilizes the cell cycle regulator Cyclin A1, controlling cell proliferation.
4. Nuclear Functions of DUBs
DUBs regulate signaling pathways that culminate in changes to gene transcription via specific transcription factors altering stability, activity, and subcellular localizations. Besides these paramount functions, DUBs also influence chromatin structures and co-ordinate DNA repair pathways. The entire cellular genome is tightly packaged in nucleosomes, the basic units of chromatin, which undergo remodeling to accommodate DNA transcription, replication, and cell division. Two major histone proteins, H2A and H2B, commonly undergo post-translational modifications via the deubiquitinating pathways that regulate chromatin structure dynamics and gene transcription. These processes are frequently altered in different types of cancers [157][158][159][160]. DUBs such as USP3, USP22, and USP46 have been reported to act on both H2A and H2B, while MYSM1, USP16, USP21, and BAP1 act on the mono-ubiquitinated H2A, and the K63-specific DUB BRCC3 shows specificity for di-ubiquitinated H2A [158][159][160][161]. USP10 has been reported to specifically act on the H2A.Z variant [162]. USP7 is associated with several factors linked to transcription, and, along with USP11, forms a part of the E3 ligase protein regulator of cytokinesis 1 (PRC1) complex, where they function to stabilize PRC1 components by their DUB activities promoting H2A ubiquitination [163]. DUBs also influence RNA-associated proteins; for example, USP4 has been reported in mRNA splicing [164][165].
The DNA damage response (DDR) is a vital aspect of physiological processes such as homeostasis and cancer prevention and is responsible for cell cycle checkpoint activation, ensuring effective DNA damage repair [166]. Cancer development has been linked to genetic damage repair, as observed in Fanconi anemia, where increased tumor rates occur in diseases with deficient DNA repair mechanisms. USP1 has been reported as a major DUB that regulates multiple aspects of the DDR pathways, including FANCD2, CHEK1, PCNA, and DDB1 [167][168][169][170]. DDRs coordinated by ATM- and ATR-CHEK are also regulated by DUBs including USP15, USP19, USP28, and USP34, of which USP28 is reported to act on CHEK2 in response to double-strand breaks (DSBs) [171][172][173]. Several other DUBs, such as USP3, USP16, BRCC36, and OTUB1, have been implicated in the regulation of DNA repair via the RNF8/168 pathway for DSB repair [174]. USP9X is another potentially important therapeutic DUB that functions to maintain DNA replication fork stability and mediate responses at DNA damage checkpoints via claspin regulation [175]. It also affects radio-sensitivity in glioblastoma cells through MCL1-dependent as well as independent mechanisms [176].
5. Drugs Targeting DUBs in Cancer
As discussed above, the significance of DUBs as key regulators in multiple biological pathways, especially their roles in cancer, has made them attractive targets for the development of new anti-cancer strategies. The DUB inhibitors that have been developed are discussed in the upcoming section in brief and summarized in Table 1.
Table 1. Summary of DUB inhibitors in oncology.[177][178][179][180][181][182][183][184][185][186][187][188][189][190][191][192][193][194][195][196][197][198][199][200][201][202][203][204][205][206][207][208][209][210][211][212][213][214][215][216][217][218][219][220][221][222][223][224][225][226][227][228][229][230][231][232][233][234]
|
Inhibitor |
DUB Target |
Mechanism of Action |
Reference |
|
b-AP15 |
USP14, UCHL5 |
19S RP inhibitor |
[177,178] |
|
VLX1570 |
USP14, UCHL5 |
Induction of apoptosis |
[179,180] |
|
Curcumin |
UPS dysregulation |
CSN5 mediated PD-L1 inhibition |
[181,182] |
|
AC17 |
USP14, UCHL5, and POH1 |
19S RP inhibitor |
[183] |
|
Δ12-PGJ2 |
UCHL1, UCHL3 |
Oxidative stress |
[184] |
|
15Δ-PGJ2 |
UCHL1 |
Inhibition of hydrolase activity |
[185–187] |
|
F6 (NSC 632839) |
USP2, USP7, and SENP2 deSUMOylase |
Induction of apoptosis |
[188,189] |
|
G5 compounds |
Broad-spectrum DUB inhibition |
Induction of apoptosis |
[190] |
|
RA-9, RA-14, AM146 |
UCHL1, UCHL3, USP2, USP5, and USP8 |
DUB active site targeting |
[191,192] |
|
WP1130 |
USP9X, USP5, USP14, and UCHL5 |
DUB active site targeting |
[193–196] |
|
P022077 |
USP7, USP47, USP10 |
Induction of p53-mediated apoptosis |
[197,198] |
|
Compound14 |
USP7, USP47 |
Induction of p53-mediated apoptosis |
[199] |
|
HBX41108 |
USP7, Non-specific DUB inhibitor |
Uncompetitive reversible p53-mediated inhibition |
[196,200] |
|
HBX19818 |
USP7, USP10 |
DUB active-site-targeted nucleophilic attacks |
[201,202] |
|
HBX28258 |
USP7 |
DUB active-site-targeted nucleophilic attacks |
[201] |
|
P5091 |
USP7 |
Cytotoxicity via HDM2-p21 signaling and p53 |
[203,204] |
|
XL177A |
USP7 |
Upregulation of p53 transcriptional targets |
[205] |
|
ADC-01, ADC-03 |
USP7 |
Unknown |
[206] |
|
GNE-6640, GNE- 6776 |
USP7 |
Attenuate Ub binding at catalytic cysteine |
[207] |
|
FT671, FT827 |
USP7 |
Target dynamic pocket at catalytic site |
[208] |
|
ML364 |
USP2, USP8 |
Reversible active site inhibition |
[209] |
|
5-(2-thienyl)-3-isoxazoles |
USP2a |
Inhibit catalytic binding site |
[210] |
|
Vialinin A |
USP5/isopeptidase T (isoT) and UCH-L1 |
Competitive active site inhibition |
[211,212] |
|
9-Ethyloxyimino-9H-indeno[1,2-b]pyrazine-2,3-dicarbonitrile |
USP8 |
Unknown |
[213,214] |
|
9-oxo-9 H-indeno[1,2-b]pyrazine-2,3-dicarbonitrile |
USP8 |
O-alkyloxime moieties at position 9 of the tricyclic scaffold |
[213,214] |
|
IU1 |
USP14 |
Suppression of Ub chain trimming |
[215,216] |
|
[1,2,3] Triazolo [4,5-d] pyrimidine |
USP28 |
Benzyl group attached to triazole ring required for its activity |
[217] |
|
3-Amino-2-keto-7H-thieno[2,3-b]pyridin-6- one derivative |
UCHL1 |
Carboxylate group at 5-position and 6-pyridone ring responsible for inhibition |
[218] |
|
LDN57444, LDN91946 |
UCHL1 |
Reversible, active-site-directed inhibitors |
[219,220] |
|
Pimozide |
USP1, Non-specific DUB inhibitor |
Reversible, non-competitive inhibition |
[196,221] |
|
ML323 |
USP1/UAF1 |
Reversible, non-active-site-targeting inhibitor |
[222,223] |
|
Mitoxantrone |
USP11 |
USP11 inhibition via unknown mechanism |
[224] |
|
Eeyarestatin-1 |
Ataxin-3 |
Unknown |
[225] |
|
GSK2643943A |
USP20 |
Unknown |
[226] |
|
PR-619 |
Broad-spectrum DUB inhibitor |
Accumulation of 26S proteasomal complexes |
[227] |
|
Betulinic acid |
Broad-spectrum DUB inhibitor |
Enhanced degradation of proliferation and pro-survival proteins |
[228] |
|
TCID |
UCHL3 |
Unknown |
[229] |
|
EOAI3402143 (G9) |
USP9X/USP24,USP5 |
Oxidative-stress-mediated apoptosis |
[230,231] |
|
Spautin-1 |
USP10, USP13 |
Inhibits autophagy via Beclin1 in Vps34 complexes |
[232] |
|
C527 |
USP1/UAF1 |
Degradation of ID1 causing p21 upregulation and cell cycle arrest |
[233] |
|
15-oxospiramilactone (S3) |
USP30 |
Promotes mitochondrial fusion via Mfn1/2 ubiquitination |
[234] |
5.1. DUB Inhibition by Compounds Containing Michael Acceptors
Michael-acceptor-containing compounds have inhibitory effects on cysteine DUBs, as they can potentially form covalent adducts with free thiols in active sites [191]. Chalcone compounds and cyclopentenone prostaglandins (PGs) of the PGJ2 class are examples of Michael-acceptor-containing compounds that are discussed below.
b-AP15: USP14 and UCHL5 are highly expressed in MM cells when compared to normal plasma cells. b-AP15 has been identified as a 19S regulatory particle (RP) inhibitor that can selectively block USP14 and UCHL5 activities without proteasomal activity inhibition [177][178]. b-AP15 has been shown to decrease the viability of patient MM cells as well as MM cell lines and, even in the presence of bone marrow stromal cells, inhibits MM cell proliferation and overcomes bortezomib resistance [177]. b-AP15 was also found to induce apoptosis in ovarian cancer cell lines by inhibiting UCHL5 and suppressing TP53-mutants, thereby regulating TGF-β signaling [178].
VLX1570: VLX1570 is an analog of b-AP15 that specifically inhibits USP14 and UCHL5 with improved solubility and potency [179]. It acts via the disruption of the ubiquitin proteasome degradation pathway, leading to an increase in poly-ubiquitinated proteins, inducible forms of chaperone Hsp70 and other markers characteristic of proteotoxic stress, ER stress, and oxidative stress leading to apoptosis [179][180].
Curcumin (diferuloylmethane): Curcumin is a natural polyphenolic compound extracted from the spice turmeric and reported to lead to ubiquitin proteasomal system dysregulation [181][182]. NF-κB p65 induces COP9 signalosome 5 (CSN5), required for PD-L1 stabilization via TNF-α in cancer cells. Curcumin also inhibits CSN5, diminishing PD-L1 expression in cancer cells and sensitizing cancer cells to anti-CTLA4 therapy [235]. In addition, curcumin exhibits anti-inflammatory, anti-oxidative, and anti-proliferative properties via the modulation of multiple cellular machineries[236].
AC17: AC17 is a 4-arylidene curcumin analog, developed as a 19S RP inhibitor, that acts as an irreversible USP14, UCHL5, and POH1 inhibitor, resulting in NF-κB pathway inhibition and reactivation of p53 [183]. AC17 causes a rapid and marked accumulation of poly-ubiquitinated proteins in A549 and NCl-H1299 cells, but the exact mechanism by which it induces the accumulation of ubiquitinated proteins is not known[183].
Δ12-PGJ2 and 15Δ-PGJ2: Δ12-PGJ2 and 15Δ-PGJ2 are inhibitors of the UCH family DUBs. Δ12-PGJ2 inhibits UCHL1 and UCHL3 with approximately 3.5 and 8.1 µM Ki, respectively, while 15Δ-PGJ2 only targets UCHL1 [184][185][186]. Δ12-PGJ2 has been reported to induce oxidative stress by causing a decrease in mitochondrial membrane potential, glutathione and glutathione peroxidases, and production of protein-bound lipid peroxidation products such as acrolein [184]. 15Δ-PGJ2 inhibits the activity of UCHL1 by affecting its structure and was the first protein reported to be oxidized in cells exposed to 15Δ-PGJ2 [185][186][187].
F6 (NSC 632839) and G5 compounds: These compounds were developed as broad-spectrum DUB inhibitors holding the same pharmacophore as Δ12-PGJ2 and induce apoptosis via accumulation of poly-ubiquitinated proteins [188]. G5 compounds induce apoptosis at low concentrations (~1 μM) and necrosis at higher concentrations (~10 μM) [190], whereas F6 inhibits the activity of USP2, USP7, and SENP2 deSUMOylase [189].
RA-9, RA-14, and AM146: These chalcone-based derivatives exhibit anti-cancer activities via DUB targeting without affecting the 20S proteasome catalytic core [191]. These inhibitors act on UCHL1, UCHL3, USP5, USP8, and USP2, which are known regulators of important cell survival and proliferation factors [191].
WP1130: WP1130 is the best-known USP9X inhibitor and has been reported to affect other DUBs (USP5, USP14, and UCHL5) [193][195]. WP1130 was identified in a screen for Janus kinase 2 (JAK2), and mass spectrometry shows a reversible covalent mechanism of action [196]. Inhibition of tumor-activated DUBs by WP1130 results in the downregulation of anti-apoptotic and upregulation of pro-apoptotic proteins such as MCL-1 and p53 and has also been reported to block the autophagic flux by ULK1 activity inhibition [193][194][195].
5.2. Other Small Molecule DUB Inhibitors
Numerous small molecules capable of inhibiting the activities of deubiquitinating enzymes have been investigated over the years. For example, USP7, or HAUSP, has been shown to have multifaceted roles in the cell cycle, DNA repair, chromatin remodeling, and epigenetic regulation. Many inhibitors have been developed to target USP7, such as P22077, HBX41108, HBX19818, HBX28258, P5091, and compound 14 [199][204][227].
Compound 14 and P22077: These compounds also inhibit USP47 along with USP7 [199][227]. Treatment of cells with Compound 14 leads to cell penetration and modulation of p53 and p21 as well as modestly accelerating polβ protein degradation in HeLa cells, confirming their role as dual USP7/USP47 inhibitors [199]. P22077 predominately inhibits USP7, showing an initial decrease in MDM2 levels followed by increased p53 and p21 levels, thereby regulating the p53 apoptotic pathway [227]. Orthotropic neuroblastoma mouse models showed an inhibition of xenograft growth in studies using P22077 [237]. P22077 also inhibits USP10 while playing a role in the degradation of oncogenic fms-related TK 3 (FLT3) kinase [197][198].
HBX family compounds: HBX41108 was the first sub-micromolar USP7 inhibitor reported but was later shown to be a non-specific DUB inhibitor [196,200]. HBX19818 and HBX28258 have been subsequently developed as more selective amidotetrahydroacridine derivatives, however, with lower potency [201]. HBX19818 shows preferential binding to the catalytic Cys residue of USP7 over other cysteinyl groups, stabilizing p53 and promoting G1 arrest and apoptosis in cells [201]. HBX19818 was recently reported to be a USP10 inhibitor that degrades spleen TKs and FLT3, resulting in the death of acute myeloid leukemia cells [202].
P5091: P5091, as with P22077, is another non-specific USP7 inhibitor from the Progenra’s thiophene chemical series [203][204]. P5091 stabilizes p53 and inhibits tumor growth in multiple myeloma cells and was found to be well tolerated in animal models while prolonging survival [204]. P5091 suppresses ovarian cancers harboring wild-type or mutant p53 genes and can effectively inhibit cell growth and induce both necrosis and apoptosis [238].
XL177A: XL177A is a selective (proteome-wide), highly potent (sub-nanomolar), and irreversible USP7 inhibitor. Transcriptome-wide analysis shows the specific upregulation of p53 transcriptional targets by XL177A [205]. Two pediatric cancers, malignant rhabdoid tumor (MRT) and Ewing sarcoma, that show sensitivity to other p53-dependent cytotoxic drugs, displayed more sensitivity to XL177A [205].
Other USP7 inhibitors: Almac Discovery and Genentech have reported fragment-based screening techniques that provide hits as starting points for USP7 discovery programs, and ADC-01 is one such hit. Utilizing X-ray crystallography techniques, they have produced a highly selective USP7 inhibitor called ADC-03 [206]. GNE-6640 and GNE-6776 are USP7 inhibitors discovered using nuclear magnetic resonance (NMR)-based screening. They enhance cytotoxicity and induce tumor cell death along with chemotherapeutic agents and targeted compounds such as PIM kinase inhibitors [207]. Simultaneously, another group has also identified two compounds, FT671 and FT827, as high-affinity USP7 inhibitors that target a dynamic pocket near the catalytic center of the auto-inhibited apo form of USP7 [208].
ML364, 5-(2-thienyl)-3-isoxazoles derivatives: USP2a is an important target for inhibition that could aid cancer therapy. ML364 has been identified as a potential small molecule inhibitor of USP2 with an IC50 of 1.1 µM, which induces cellular cyclin D1 degradation leading to cell-cycle arrest in HCT116 cancer cell lines and also has a lower affinity to USP8 [209]. ML364 selectively inhibits USP2 and thereby increases mitochondrial ROS levels, modulating the morphology and membrane potential of the mitochondria [239]. Two other small molecule inhibitors of USP2a, derivatives of 5-(2-thienyl)-3-isoxazoles, have been identified using NMR-based fragment screening and biophysical binding assays [210].
Vialinin A: Vialinin A, a non-specific USP4 inhibitor with an IC50 value of 1.5 µM, also targets the catalytic activity of USP5/isopeptidase T (isoT) and UCH-L1 [211]. Vialinin A plays a role in the prevention of VEGF-induced neovascularization, and research suggests that its natural anti-oxidative properties could be developed as anti-angiogenic agents in cancer therapy [212].
9-Oxo-9H-indeno[1,2-b]pyrazine-2,3-dicarbonitrile and analogs: 9-Oxo-9H-indeno[1,2-b]pyrazine-2,3-dicarbonitrile and its analogs have been identified by high-throughput screens as selective USP8 inhibitors that show anti-proliferative and pro-apoptotic activities in various cancer cell lines [213][214]. 9-Ethyloxyimino-9H-indeno [1,2-b]pyrazine-2,3-dicarbonitrile has been identified as an USP8 inhibitor that is structurally similar to USP7 inhibitors, and derivatives of these compounds have shown to be efficacious in mouse lung cancer models [213][214].
IU1 (1-[1-(4-fluorophenyl)-2,5-dimethylpyrrol-3-yl]-2-pyrrolidin-1-ylethanone): IU1 is an active-site-directed thiol protease small molecule inhibitor that selectively inhibits Usp14 [215]. It has been recently reported that IU1-mediated inhibition of USP14 results in the increased ubiquitination of constitutive photo-morphogenesis 9 (COP9) signalosome subunit 5 (COPS5), a key negative p53 regulator, resulting in tumor regression of autochthonous T-lymphomas and sarcomas in p53-deficient mice without affecting normal tissues [216].
[1,2,3] Triazolo [4,5-d] pyrimidine and derivatives: [1,2,3] Triazolo [4,5-d] pyrimidine and derivatives have been identified as inhibitors of USP28, especially compound 19, which has an IC50 value of 1.1 µM/L and a Kd value of 40 nM/L [217]. This novel inhibitor directly inhibits USP28 and induces the inhibition and degradation of the cell cycle, cell proliferation, and EMT progress in gastric cell lines [217].
3-Amino-2-keto-7H-thieno[2,3-b]pyridin-6-one derivative: 3-Amino-2-keto-7H-thieno[2,3-b]pyridin-6-one derivative is a UCHL1-targeting small molecule, discovered as a moderately potent non-competitive inhibitor that works by binding only to the Michaelis complex and not to free enzyme [218].
Isatin O-acyl oximes: Isatin O-acyl oximes are UCHL1-targeting inhibitors that can selectively inhibit UCHL1 in preference to its systemic isoform UCHL3 [219]. LDN57444 and LDN91946 are examples of UCHL1-inhibiting isatin O-acyl oximes [219]. Inhibition of UCHL1 activity using LDN5744 or its nanoparticle formulation LDN-POx in vitro shows inhibition of exosome secretions and reduced levels of pro-metastatic factors in exosomal fractions while suppressing the motility of metastatic squamous carcinoma cells and nasopharyngeal cells expressing EBV pro-metastatic latent membrane protein 1 (LMP1) [220]. LDN and LDN-POx treatment also showed decreased carcinoma cell adhesion, reduced pro-metastatic markers, and inhibition of extracellular vesicle-mediated transfer of invasive factor LMP1 [220].
Pimozide: Pimozide was initially developed as a selective USP1 inhibitor with sub-micromolar potency, shown to sensitize platinum-resistant NSCLC cells and promote PCNA and FANCD2 mono-ubiquitination [221]. Although pimozide studies show intended results, DUB selectivity profiling suggests non-specific behavior of the drug [196].
ML323: ML323 is another notable USP1/UAF1 complex inhibitor containing a selective pyrimidine core compound and has been shown to allosterically block UAF1 and USP1 complex formation, potentiate cisplatin cytotoxicity, and increase PCNA and FANCD2 mono-ubiquitination in cells [222][223]. However, not much progress has been made in advancing USP1 inhibitors to clinical settings.
Mitoxantrone: Mitoxantrone was previously used for the treatment of acute myeloid leukemia, hormone-refractory prostate cancer, and multiple sclerosis and is the only reported topoisomerase inhibitor of USP11 that functions via an unknown mechanism [224].
Eeyarestatin-1 and GSK2643943A: Eeyarestatin-1 (Eer1) has been reported to specifically target DUBs associated with p97/VCP, such as Ataxin-3 [225]. A potent USP20 inhibitor identified by GSK, GSK2643943A, showed an IC50 of 160 nM [226].
6. Drug Development Challenges and Ongoing Clinical Trials
Dramatic advancements in the field of UPS have greatly enhanced our understanding of the functions and mechanisms of this system. Previously, researchers focused on identifying compounds that disrupt E3 ligase and its substrate interactions, which can be intrinsically more difficult to achieve than searching for small molecule catalytic blockers. The challenges faced by researchers include identifying potent compounds that could selectively target DUBs at their catalytic pockets. The second challenge faced is due to Ub transfer via reactive thiol groups by DUBs, which interferes with the screening of DUB inhibitors. This is because the standard assays that have been used to identify inhibitors are limited by non-selective redox or alkylating false positives [240]. The complex mechanisms of DUBs as they alternate between active and non-active conformations also present a challenge when designing predictive biochemical assays and developing drug-like compounds [241][242]. Finally, the Ub specificity between the DUBs and the target proteins poses a challenge to optimizing the likelihood of identifying genuine inhibitors.
Table 2. Inhibitors of the UPS.[243][244][245][246][247][248][249][250][251][252][253][254][255][256][257][258][259][260][261][262][263][264][265][266][267][268][269][270][271][272][273][274][275][276][277][278]
|
Inhibitor |
Target |
Reference |
|
PYR-41 |
E1 enzyme |
[248,249] |
|
MLN7243 |
E1 enzyme |
[250] |
|
MLN4924 |
E1 enzyme |
[251] |
|
Compound 4b |
E1 enzyme |
[252] |
|
PYZD-4409 |
E1 enzyme |
[253] |
|
Leucettamol A |
E2 enzyme |
[254] |
|
Manadosterols A and B |
E2 enzyme |
[255] |
|
CC0651 |
E2 enzyme |
[256] |
|
Nutlins and derivatives, RITA, MI-219, Syl-155, MI-63, PRIMA-1, HLI98, HLI373, MEL23 and MEL24, ATSP-7041, NSC207895 |
Mdm2/Mdmx/p53-mediated E3 ligase enzyme |
[257–267] |
|
Oridonin, SCF-12, ZL25, Compound A, Erioflorin, GS143, SMER3, TAME, Apcin |
SCF E3 ligase |
[268–276] |
|
Bortezomib, CEP-18770, Carfilzomib (PR-171), ONX-0912, PR-047, MLN9708 and MLN2238 (Ixazomib), Marizomib (NPI-0052) |
Proteasomal inhibitors |
[277–283] |
Targeting upstream regulators of the UPS, such as the E1, E2, and E3 enzymes, provides another level of control over the ubiquitination–deubiquitination system. Even targeting the proteasome has proven a successful clinical therapy, highlighted by the success of some FDA-approved inhibitors such as bortezomib [243], carfilzomib [244], oprozomib (ONX0912) [245], and ixazomib [246]. Inhibitors that have been identified to target the UPS and are under pre-clinical trials are summarized in Table 2.
However, these drugs have shortcomings, such as fatigue, asthenia, drug resistance (bortezomib), and cardiovascular complications (carfilzomib), and need further optimization. Understanding the mechanism of E3 ligase specificity toward E2 enzymes and substrates and the trigger of the proteasomal degradation pathway due to lysine specificity could help explain the underlying mechanisms via structure–function studies. These avenues could open new targeting strategies for the development of highly specific and potent inhibitors. Despite the growing attractiveness of DUBs as therapeutic targets and the advancements in the field of DUB discovery and target identification, only a handful of DUB inhibitors, such as VLX1570, have advanced through clinical trials for cancer therapy. However, these phase I trials had to be prematurely terminated due to severe toxicity [279][280][281][282][283][284]. Complete information regarding clinical trials can be accessed using the clinical trial ID [285] and the promising DUB inhibitors undergoing clinical trials are reviewed in this section.
The naturally occurring compound curcumin, which has been reported to have UPS dysregulation properties, has been extensively studied in clinical settings, especially in cancers [285]. Samsung Medical Center completed a placebo-controlled, double-blind, randomized trial in 107 male participants using curcumin in 2017 (NCT03211104). They aimed at establishing whether curcumin influenced the duration of treatment interruption and rate of prostatic specific antigen (PSA) progression, compared with placebo, among men with prostate cancer receiving intermittent androgen deprivation therapy. A phase II trial initiated by Emory University (NCT02944578) in 2016 is attempting to evaluate the biomolecular effects of curcumin capsules in high-grade squamous intraepithelial lesion (HSIL) cervical neoplasia. This drug is being tested in 40 women and has been postulated to affect cancerous cells by modulating various cellular pathways and altering the effect of HPV on tissue cells. This study aims at exploring the effect of curcumin as a potential medical treatment in HIV-infected women with HSIL lesions of the cervix. A recent study proposed by the Medical University of South Carolina (NCT03980509) in 2019 is recruiting to determine the effect of oral administration of curcumin in primary breast cancer tumors in 20 participants. This phase I trial will use oral administration of curcumin to check DNA-fragmentation-related apoptosis and cell proliferation (Ki67) in primary tumors of breast cancer patients.
Mitoxantrone is an FDA-approved drug that reportedly inhibits USP11. Numerous phase I/II clinical trials are currently being conducted using mitoxantrone to target various diseases, including neoplasms, relapsed/refractory acute myeloid leukemia, multiple sclerosis, advanced recurrent or metastatic breast cancer, neuromyelitis optica, and more [286][287]. The interventional study NCT02043756, completed in 2014, tested the pharmacokinetics, toxicity, and maximum tolerated dose of mitoxantrone hydrochloride (plm60-s) liposome injections in 20 participants with solid tumors. This study reported that a dose of up to 18 mg/m(2) of plm60-s had potential efficacy and was well tolerated [287]. A currently active phase II trial, initiated in December, 2018 is being conducted by CSPC ZhongQi Pharmaceutical Technology Co., Ltd. (NCT03776279). This trial aims to evaluate the safety and efficiency of mitoxantrone hydrochloride liposome injections in 106 participants with relapsed/refractory peripheral T-cell and NK/T-cell lymphoma. Michael Boyiadzis’ group, from the University of Pittsburgh (NCT03839446), initiated a phase II study in 2019 to examine the efficiency and toxicity of mitoxantrone in combination with etoposide and gemtuzumab ozogamicin (MEGO) in acute myeloid leukemia patients. This study is aimed towards patients who did not respond to first-line induction therapy to examine the efficacy and toxicity of this combinational therapy.
Another important DUB inhibitor that has advanced to clinical trials is pimozide, which is currently in a phase II trial in patients with amyotrophic lateral sclerosis (ALS). The University of Calgary is actively conducting two clinical trials using pimozide (NCT02463825, NCT03272503). The first study, initiated in 2015, is a phase II trial that evaluates the effect of pimozide in 25 patients with neuromuscular junction transmission dysfunction due to amyotrophic lateral sclerosis (ALS) (NCT02463825). The second study, initiated in 2017, is a phase II placebo-controlled randomized trial using 100 ALS patients (NCT03272503). This study aims to test the effect of pimozide in slowing the progression of ALS. Successful clinical trials were also conducted in 2019 using betulinic acid, hypothesized to demonstrate anxiolytic and/or stress-reducing properties (NCT03904511). A betulinic-acid-containing plant extract called the Souroubea-Platanus preparation was administered to 45 healthy participants to study its safety, tolerability, and behavioral effects in healthy volunteers (NCT03904511). A complete list of inhibitors targeting DUBs and UPS enzyme components, along with their associated diseases, which are in different stages of clinical trials, is summarized in Table 3.
Table 3. Clinical status and disease association of UPS system inhibitors.[288][289][290][291][292][293][294][295][296][297][298][299][295][296][297][298][299][300][301][302][303][304][305][306][307][308][309][310][311][312][313][314][315][316][317][318][319][320][321][322][323][324]
|
Inhibitor |
Target |
Clinical Status |
Cancer Type and Disease |
Reference/ Clinical Trial ID |
|
b-AP15 |
USP14, UCHL5 |
Patent (ID: W0201305869)Preclinical |
Acute myeloid leukemia, Multiple myeloma, Mantle cell lymphoma, Neuroblastoma, Prostate cancer, Colon cancer, Ovarian cancer, Breast cancer, Large B cell lymphoma |
[177,249,288–292] |
|
VLX1570 |
USP14, UCHL5 |
Phase I/II, prematurely ended |
Acute myeloid leukemia, Multiple myeloma, Mantle cell lymphoma, Neuroblastoma, Prostate cancer, Colon cancer, Ovarian cancer, Breast cancer, Large B cell lymphoma |
[293,294] |
|
Curcumin |
UPS dysregulation |
Phase I/II, trials going |
Prostate cancer, Lung cancer, Crohn’s disease, Colorectal cancer, Head and neck cancer, Breast cancer, Colonic cancer, Metastasis, Advanced cancers, Chronic obstructive pulmonary disease, Metabolic syndrome |
[295,296] |
|
WP1130 |
USP9X, USP5, USP14 and UCHL5 |
Patent (ID: W02012204527 A2) Preclinical |
Acute myeloid leukemia, Prostate cancer, Colon cancer, Lung cancer, Hepatocellular carcinoma, Mesothelioma, Chronic myelogenous leukemia |
[193,297–299] |
|
Pimozide |
USP1 |
Phase I/IV trials in schizophrenia Phase II trials in Amyotrophic Lateral Sclerosis (ALS) |
Schizophrenia, Psychotic Disorders, Tourette Syndrome, Amyotrophic lateral sclerosis (ALS) |
NCT02463825, NCT03272503, NCT00374244 |
|
Mitoxantrone |
USP11 |
FDA approved |
Acute myeloid leukemia, Neoplasms, Breast cancer, Acute myelogenous leukemia, Lymphoblastic lymphoma, Diffuse large B-cell lymphoma, Burkitt lymphoma/Leukemia |
[287,300,301] |
|
Betulinic acid |
Broad spectrum DUB inhibitor |
Phase I |
Anxiety, Stress, Psychological disorders |
NCT03904511 |
|
MLN7243 |
E1 enzyme |
Phase I |
Advanced malignant solid tumors, Myelodysplastic syndrome, Recurrent/refractory acute myeloid leukemia, Refractory chronic myelomonocytic leukemia, Refractory high-risk myelodysplastic syndrome |
NCT03816319 |
|
MLN4924 |
E1 enzyme |
Phase I, completed |
Acute myelogenous leukemia
|
[302,303]
|
|
Compound 4b |
E1 enzyme |
Phase I, completed |
Neoplasms, Epilepsy, HIV infections
|
[304] |
|
Bortezomib |
Proteasomal inhibitor |
FDA approved |
Multiple myeloma, Mantle cell lymphoma, Leukemia, Neuroblastoma, Head and neck cancer, Thyroid carcinoma, Hepatocellular carcinoma, Amyloidosis, Cold agglutinin disease, Hemolytic anemia |
[305–310] |
|
Carfilzomib |
Proteasomal inhibitor |
FDA approved |
Multiple myeloma, Relapsed and/or refractory multiple myeloma, Lymphoma, Leukemia Lung cancer, Thyroid carcinoma, Refractory renal cell carcinoma, Pulmonary hypertension |
[311–314] |
|
Ixazomib |
Proteasomal inhibitor |
FDA approved |
Multiple myeloma, Relapsed and/or refractory multiple myeloma, Lymphoma, Leukemia, Breast cancer, Glioblastoma, Renal cell carcinoma, Bladder cancer, Hodgkin and T cell lymphoma, HIV, Lupus, Kidney diseases |
[315–318] |
|
Marizomib |
Proteasomal inhibitor |
Phase I/II/III ongoing |
Multiple myeloma, Relapsed and/or refractory multiple myeloma, Lymphoma, Glioblastoma, Pancreatic cancer, Melanoma, |
[319–321]
|
|
ONX-0912 |
Proteasomal inhibitor |
Phase I/II, completed |
Multiple myeloma, Relapsed and/or refractory multiple myeloma, Hepatocellular carcinoma, Non-central-nervous-system malignancies |
[322–324] |
7. Conclusion
The regulation of many critical proteins and cellular signaling events by DUBs modulates homeostasis and cell fate. Dramatic advancements in understanding of the ubiquitin system and identification of the crystal structures of various DUBs have paved the way for new possibilities in drug discovery and treatment of proteopathies, neurological disorders, and cancer. Parallel to the research focused on discovering different facets of DUB activity, the increasing number of inhibitors identified against this system have proved to be efficacious and selective in treating several disorders, including cancer. These inhibitors targeting the UPS system components are summarized in Table 3.
Despite our growing understanding of DUB biology, considerable work needs to be done to apply DUBs in clinical research. In-depth studies are therefore required to understand their natural regulatory mechanisms. In addition, identification of candidate pathways and targets in a cell that may assist DUB pharmacology also requires attention. Furthermore, pharmacodynamic effects of DUBs can be explored for possible avenues in clinical research. Besides DUB inhibitors, drugs enhancing DUB activities or expression should also be considered for further research. Another gray area in the field of DUBs is the number of targets regulated by a single DUB and vice versa. This raises a new challenge in itself as the inhibitors developed could have severe adverse effects by affecting non-target pathways. A combination of technologies, such as genomics, proteomics and structural analysis of DUBs, and an examination of their interactions with specific targets, combined with drug delivery strategies, could help advance the field of DUB inhibitors and accelerate them into the clinical setting. Combination therapies consisting of already approved proteasomal inhibitors along with novel DUB inhibitors could help abate the side effects of these drugs and also provide an interesting line of investigation in current research.
Abbreviations:
|
DUBs |
Deubiquitinating enzymes |
|
UPS |
Ubiquitin proteasome system |
|
UCHs |
Ubiquitin C-terminal hydrolases |
|
OTUs |
Ovarian tumor proteases |
|
MJDs |
Josephin or Machado–Joseph disease protein domain proteases |
|
JAMM |
Jab1/MPN domain-associated metalloisopeptidase domain proteins |
|
MCPIP |
Monocyte chemotactic protein-induced protein family |
|
MINDY |
Motif interacting with Ub-containing novel DUB family |
|
ZUFSPs |
Zn-finger and UFSP domain proteins |
|
GWAS |
Genome-wide association studies |
|
RTKs |
Receptor tyrosine kinases |
|
Ub |
Ubiquitin |
|
BAP1 |
BRCA1-associated protein 1 |
|
MM |
Multiple myelomas |
|
MCL |
Myeloid cell leukemia |
|
CDH11 |
Cadherin11 gene |
|
NF-κB |
Nuclear factor-κB |
|
IKK |
IκB kinase |
|
TPA |
12-O-tetradecanoylphorbol-13 acetate |
|
TNFAIP3 |
tumor necrosis factor, alpha-induced protein 3 |
|
HCF-1 |
Host cell factor-1 |
|
TGF-β |
Transforming growth factor-β |
|
AIF |
Apoptosis-inducing factor |
|
FASN |
Fatty acid synthase |
|
EGFR |
Epidermal growth factor receptor |
|
MEFs |
Mouse epithelial fibroblasts |
|
FOXO4 |
Forkhead box protein O4 |
|
PTEN |
Phosphatase and tensin homolog |
|
NSCLC |
Non-small-cell lung cancer |
|
LSD1 |
Lysine-specific demethylase1 |
|
TP53BP1 |
TP53-binding protein 1 |
|
MCPIP1 |
MCP-induced protein 1 |
|
TK |
tyrosine kinase |
|
PCP |
Planar cell polarity |
|
EMT |
epithelial-to-mesenchymal transition |
|
R-SMAD |
Receptor-regulated SMADs |
|
DDR |
DNA damage response |
|
19S RP |
19S regulatory particle |
|
CSN5 |
COP9 signalosome 5 |
|
JAK2 |
Janus kinase 2 |
|
FLT3 |
Fms-related tyrosine kinase 3 |
|
CYLD |
Cylindromatosis |
|
APC |
Anaphase-promoting complex |
|
Plk1 |
polo-like kinase 1 |
References
- Stadler, Z.K.; Thom, P.; Robson, M.; Weitzel, J.N.; Kauff, N.; Hurley, K.E.; Devlin, V.; Gold, B.; Klein, R.J.; Offit, K. Genome-Wide Association Studies of Cancer. J. Clin. Oncol. 2010, 28, 4255–4267, doi:10.1200/jco.2009.25.7816.
- Sud, A.; Kinnersley, B.; Houlston, R. Genome-wide association studies of cancer: current insights and future perspectives. Nat. Rev. Cancer 2017, 17, 692–704, doi:10.1038/nrc.2017.82.
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 789–799, doi:10.1038/nm1087.
- Shortt, J.; Johnstone, R.W. Oncogenes in Cell Survival and Cell Death. Cold Spring Harb. Perspect. Biol. 2012, 4, a009829, doi:10.1101/cshperspect.a009829.
- Küppers, R.; Dalla-Favera, R. Mechanisms of chromosomal translocations in B cell lymphomas. Oncogene 2001, 20, 5580–5594, doi:10.1038/sj.onc.1204640.
- Chial, H. Tumor suppressor (TS) genes and the two-hit hypothesis. Nat. Educ. 2008, 1, 177.
- A Jeggo, P.; Pearl, L.; Carr, A.M. DNA repair, genome stability and cancer: a historical perspective. Nat. Rev. Cancer 2015, 16, 35–42, doi:10.1038/nrc.2015.4.
- Pearl, L.; Schierz, A.C.; Ward, S.E.; Al-Lazikani, B.; Pearl, F.M. Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer 2015, 15, 166–180, doi:10.1038/nrc3891.
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339, doi:10.1038/nature12634.
- Kan, Z.; Jaiswal, B.S.; Stinson, J.; Janakiraman, V.; Bhatt, D.; Stern, H.M.; Yue, P.; Haverty, P.M.; Bourgon, R.; Zheng, J.; et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 2010, 466, 869–873, doi:10.1038/nature09208.
- Lopez-Otin, C.; Bond, J.S. Proteases: Multifunctional enzymes in life and disease. J. Biol. Chem. 2008, 283, 30433–30437, doi:10.1074/jbc.R800035200.
- Mason, S.D.; Joyce, J.A. Proteolytic networks in cancer. Trends Cell Biol. 2011, 21, 228–237, doi:10.1016/j.tcb.2010.12.002.
- Turk, B. Targeting proteases: Successes, failures and future prospects. Nat. Rev. Drug Discov. 2006, 5, 785–799, doi:10.1038/nrd2092.
- Drag, M.; Salvesen, G.S. Emerging principles in protease-based drug discovery. Nat. Rev. Drug Discov. 2010, 9, 690–701, doi:10.1038/nrd3053.
- Pérez-Silva, J.G.; Español, Y.; Velasco, G.; Quesada, V. The Degradome database: expanding roles of mammalian proteases in life and disease. Nucleic Acids Res. 2015, 44, D351–D355, doi:10.1093/nar/gkv1201.
- Reyes-Turcu, F.E.; Ventii, K.H.; Wilkinson, K.D. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 2009, 78, 363–397, doi:10.1146/annurev.biochem.78.082307.091526.
- Komander, D.; Clague, M.J.; Urbe, S. Breaking the chains: structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 2009, 10, 550–563, doi:10.1038/nrm2731.
- Kaushal, K.; Antao, A.M.; Kim, K.-S.; Ramakrishna, S. Deubiquitinating enzymes in cancer stem cells: functions and targeted inhibition for cancer therapy. Drug Discov. Today 2018, 23, 1974–1982, doi:10.1016/j.drudis.2018.05.035.
- Hershko, A.; Heller, H.; Elias, S.; Ciechanover, A. Components of ubiquitin-protein ligase system. Resolution, affinity purification, and role in protein breakdown. J. Biol. Chem. 1983, 258, 8206–8214.
- Chen, Z.J.; Sun, L.J. Nonproteolytic Functions of Ubiquitin in Cell Signaling. Mol. Cell 2009, 33, 275–286, doi:10.1016/j.molcel.2009.01.014.
- Ikeda, F.; Dikic, I. Atypical ubiquitin chains: new molecular signals. EMBO Rep. 2008, 9, 536–542, doi:10.1038/embor.2008.93.
- Dammer, E.B.; Na, C.H.; Xu, P.; Seyfried, N.T.; Duong, D.M.; Cheng, N.; Gearing, M.; Rees, H.; Lah, J.J.; Levey, A.I.; et al. Polyubiquitin linkage profiles in three models of proteolytic stress suggest the etiology of alzheimer disease. J. Biol. Chem. 2011, 286, 10457–10465, doi:10.1074/jbc.M110.149633.
- Ohtake, F.; Tsuchiya, H.; Saeki, Y.; Tanaka, K. K63 ubiquitylation triggers proteasomal degradation by seeding branched ubiquitin chains. Proc. Natl. Acad. Sci. USA 2018, 115, E1401–E1408, doi:10.1073/pnas.1716673115.
- Wilkinson, K.D. Regulation of ubiquitin‐dependent processes by deubiquitinating enzymes. FASEB J. 1997, 11, 1245–1256, doi:10.1096/fasebj.11.14.9409543.
- Hymowitz, S.G.; Wertz, I.E. A20: from ubiquitin editing to tumour suppression. Nat. Rev. Cancer 2010, 10, 332–341, doi:10.1038/nrc2775.
- Amerik, A.; Hochstrasser, M. Mechanism and function of deubiquitinating enzymes. Biochim. Biophys. Acta Bioenerg. 2004, 1695, 189–207, doi:10.1016/j.bbamcr.2004.10.003.
- Rehman, S.A.A.; Kristariyanto, Y.A.; Choi, S.-Y.; Nkosi, P.J.; Weidlich, S.; Labib, K.; Hofmann, K.; Kulathu, Y. MINDY-1 is a member of an evolutionarily conserved and structurally distinct new family of deubiquitinating enzymes. Mol. Cell 2016, 63, 146–155, doi:10.1016/j.molcel.2016.05.009.
- Hermanns, T.; Pichlo, C.; Woiwode, I.; Klopffleisch, K.; Witting, K.; Ovaa, H.; Baumann, U.; Hofmann, K. A family of unconventional deubiquitinases with modular chain specificity determinants. Nat. Commun. 2018, 9, 799, doi:10.1038/s41467-018-03148-5.
- Liang, J.; Saad, Y.; Lei, T.; Wang, J.; Qi, D.; Yang, Q.; Kolattukudy, P.E.; Fu, M. MCP-induced protein 1 deubiquitinates TRAF proteins and negatively regulates JNK and NF-κB signaling. J. Exp. Med. 2010, 207, 2959–2973, doi:10.1084/jem.20092641.
- Liu, L.; Damerell, D.R.; Koukouflis, L.; Tong, Y.; Marsden, B.D.; Schapira, M. UbiHub: a data hub for the explorers of ubiquitination pathways. Bioinformatics 2019, 35, 2882–2884, doi:10.1093/bioinformatics/bty1067.
- Murali, R.; Wiesner, T.; Scolyer, R.A. Tumours associated with BAP1 mutations. Pathology 2013, 45, 116–126, doi:10.1097/pat.0b013e32835d0efb.
- Oliveira, J.L.; Chou, M.M. USP6-induced neoplasms: the biologic spectrum of aneurysmal bone cyst and nodular fasciitis. Hum. Pathol. 2014, 45, 1–11, doi:10.1016/j.humpath.2013.03.005.
- Homan, C.C.; Kumar, R.; Nguyen, L.S.; Haan, E.; Raymond, F.L.; Abidi, F.; Raynaud, M.; Schwartz, C.E.; Wood, S.A.; Gécz, J.; et al. Mutations in USP9X Are Associated with X-Linked Intellectual Disability and Disrupt Neuronal Cell Migration and Growth. Am. J. Hum. Genet. 2014, 94, 470–478, doi:10.1016/j.ajhg.2014.02.004.
- Murtaza, M.; Jolly, L.A.; Gécz, J.; Wood, S.A. La FAM fatale: USP9X in development and disease. Cell. Mol. Life Sci. 2015, 72, 2075–89, doi:10.1007/s00018-015-1851-0.
- Eichhorn, P.J.A.; Rodón, L.; Gonzàlez-Juncà, A.; Dirac, A.; Gili, M.; Martinez-Saez, E.; Aura, C.; Barba, I.; Peg, V.; Prat, A.; et al. USP15 stabilizes TGF-β receptor I and promotes oncogenesis through the activation of TGF-β signaling in glioblastoma. Nat. Med. 2012, 18, 429–435, doi:10.1038/nm.2619.
- Bignell, G.R.; Warren, W.; Seal, S.; Takahashi, M.; Rapley, E.; Barfoot, R.; Green, H.; Brown, C.; Biggs, P.J.; Lakhani, S.; et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nat. Genet. 2000, 25, 160–165, doi:10.1038/76006.
- Fraile, J.M.; Quesada, V.; Rodríguez, D.; Freije, J.M.P.; Lopez-Otin, C. Deubiquitinases in cancer: New functions and therapeutic options. Oncogene 2011, 31, 2373–2388, doi:10.1038/onc.2011.443.
- Richardson, P.G.; Hideshima, T.; Anderson, K.C. Bortezomib (PS-341): A Novel, First-in-Class Proteasome Inhibitor for the Treatment of Multiple Myeloma and Other Cancers. Cancer Control. 2003, 10, 361–369, doi:10.1177/107327480301000502.
- Chen, D.; Frezza, M.; Schmitt, S.; Kanwar, J.; Dou, Q.P. Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr. Cancer Drug Targets 2011, 11, 239–253, doi:10.2174/156800911794519752.
- D’Arcy, P.; Wang, X.; Linder, S. Deubiquitinase inhibition as a cancer therapeutic strategy. Pharmacol. Ther. 2015, 147, 32–54, doi:10.1016/j.pharmthera.2014.11.002.
- Wei, R.; Liu, X.; Yu, W.; Yang, T.; Cai, W.; Liu, J.; Huang, X.; Xu, G.-T.; Zhao, S.; Yang, J.; et al. Deubiquitinases in cancer. Oncotarget 2015, 6, 12872–12889, doi:10.18632/oncotarget.3671.
- Hussain, S.; Zhang, Y.; Galardy, P.J. DUBs and cancer: The role of deubiquitinating enzymes as oncogenes, non-oncogenes and tumor suppressors. Cell Cycle 2009, 8, 1688–1697, doi:10.4161/cc.8.11.8739.
- A Vendrell, J.; Ghayad, S.; Ben-Larbi, S.; Dumontet, C.; Mechti, N.; A Cohen, P. A20/TNFAIP3, a new estrogen-regulated gene that confers tamoxifen resistance in breast cancer cells. Oncogene 2007, 26, 4656–4667, doi:10.1038/sj.onc.1210269.
- Sun, S.C. CYLD: a tumor suppressor deubiquitinase regulating NF-κB activation and diverse biological processes. Cell Death Differ. 2010, 17, 25–34, doi: 10.1038/cdd.2009.43.
- Kon, N.; Kobayashi, Y.; Li, M.; Brooks, C.L.; Ludwig, T.; Gu, W. Inactivation of HAUSP in vivo modulates p53 function. Oncogene 2009, 29, 1270–1279, doi:10.1038/onc.2009.427.
- Oliveira, A.M.; Perez-Atayde, A.R.; Inwards, C.Y.; Medeiros, F.; Derr, V.; Hsi, B.-L.; Gebhardt, M.C.; Rosenberg, A.E.; Fletcher, J.A. USP6 and CDH11 Oncogenes Identify the Neoplastic Cell in Primary Aneurysmal Bone Cysts and Are Absent in So-Called Secondary Aneurysmal Bone Cysts. Am. J. Pathol. 2004, 165, 1773–1780, doi:10.1016/s0002-9440(10)63432-3.
- Li, L.; Yang, H.; He, Y.; Li, T.; Feng, J.; Chen, W.; Ao, L.; Shi, X.; Lin, Y.; Liu, H.; et al. Ubiquitin-Specific Protease USP6 Regulates the Stability of the c-Jun Protein. Mol. Cell. Biol. 2017, 38, e00317–e00320, doi:10.1128/mcb.00320-17.
- Shah, S.P.; Morin, R.D.; Khattra, J.; Prentice, L.; Pugh, T.J.; Burleigh, A.; Delaney, A.; Gelmon, K.; Guliany, R.; Senz, J.; et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature 2009, 461, 809–813, doi:10.1038/nature08489.
- Dubois, A.; Wilson, V.; Bourn, D.; Rajan, N. CYLD Genetic Testing for Brooke-Spiegler Syndrome, Familial Cylindromatosis and Multiple Familial Trichoepitheliomas. PLoS Curr. 2015, 7, doi:10.1371/currents.eogt.45c4e63dd43d62e12228cc5264d6a0db.
- Baud, V.; Karin, M. Is NF-κB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 2009, 8, 33–40, doi:10.1038/nrd2781.
- Trompouki, E.; Hatzivassiliou, E.; Tsichritzis, T.; Farmer, H.; Ashworth, A.; Mosialos, G. CYLD is a deubiquitinating enzyme that negatively regulates NF-κB activation by TNFR family members. Nature 2003, 424, 793–796, doi:10.1038/nature01803.
- Kovalenko, A.; Chable-Bessia, C.; Cantarella, G.; Israël, A.; Wallach, D.; Courtois, G. The tumour suppressor CYLD negatively regulates NF-κB signalling by deubiquitination. Nature 2003, 424, 801–805, doi:10.1038/nature01802.
- Brummelkamp, T.R.; Nijman, S.M.B.; Dirac, A.M.G.; Bernards, R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-κB. Nature 2003, 424, 797–801, doi:10.1038/nature01811.
- Novak, U.; Rinaldi, A.; Kwee, I.; Nandula, S.V.; Rancoita, P.M.V.; Compagno, M.; Cerri, M.; Rossi, D.; Murty, V.V.; Zucca, E.; et al. The NF-κB negative regulator TNFAIP3 (A20) is inactivated by somatic mutations and genomic deletions in marginal zone lymphomas. Blood 2009, 113, 4918–4921, doi:10.1182/blood-2008-08-174110.
- Dürkop, H.; Hirsch, B.; Hahn, C.; Foss, H.-D.; Stein, H. Differential expression and function of A20 and TRAF1 in Hodgkin lymphoma and anaplastic large cell lymphoma and their induction by CD30 stimulation. J. Pathol. 2003, 200, 214–221, doi:10.1002/path.1351.
- Song, H.Y.; Rothe, M.; Goeddel, D.V. The tumor necrosis factor-inducible zinc finger protein A20 interacts with TRAF1/TRAF2 and inhibits NF-kappaB activation. Proc. Natl. Acad. Sci. USA 1996, 93, 6721–6725, doi:10.1073/pnas.93.13.6721.
- E Jensen, D.; Proctor, M.; Marquis, S.T.; Gardner, H.P.; I Ha, S.; A Chodosh, L.; Ishov, A.M.; Tommerup, N.; Vissing, H.; Sekido, Y.; et al. BAP1: a novel ubiquitin hydrolase which binds to the BRCA1 RING finger and enhances BRCA1-mediated cell growth suppression. Oncogene 1998, 16, 1097–1112, doi:10.1038/sj.onc.1201861.
- Testa, J.R.; Cheung, M.; Pei, J.; Below, J.E.; Tan, Y.; Sementino, E.; Cox, N.J.; Dogan, A.U.; Pass, H.I.; Trusa, S.; et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat. Genet. 2011, 43, 1022–1025, doi:10.1038/ng.912.
- Wiesner, T.; Obenauf, A.C.; Murali, R.; Fried, I.; Griewank, K.; Ulz, P.; Windpassinger, C.; Wackernagel, W.; Loy, S.; Wolf, I.; et al. Germline mutations in BAP1 predispose to melanocytic tumors. Nat. Genet. 2011, 43, 1018–1021, doi:10.1038/ng.910.
- Harbour, J.W.; Onken, M.D.; Roberson, E.D.; Duan, S.; Cao, L.; Worley, L.A.; Council, M.L.; Matatall, K.A.; Helms, C.; Bowcock, A.M. Frequent Mutation of BAP1 in Metastasizing Uveal Melanomas. Science 2010, 330, 1410–1413, doi:10.1126/science.1194472.
- Guénard, F.; Brcas, I.; Labrie, Y.; Ouellette, G.; Beauparlant, C.J.; Durocher, F. Genetic sequence variations of BRCA1-interacting genes AURKA, BAP1, BARD1 and DHX9 in French Canadian Families with high risk of breast cancer. J. Hum. Genet. 2009, 54, 152–161, doi:10.1038/jhg.2009.6.
- Bott, M.; Brevet, M.; Taylor, B.S.; Shimizu, S.; Ito, T.; Wang, L.; Creaney, J.; A Lake, R.; Zakowski, M.F.; Reva, B.; et al. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat. Genet. 2011, 43, 668–672, doi:10.1038/ng.855.
- Misaghi, S.; Ottosen, S.; Izrael-Tomasevic, A.; Arnott, D.; Lamkanfi, M.; Lee, J.; Liu, J.; O’Rourke, K.; Dixit, V.M.; Wilson, A.C. Association of C-Terminal Ubiquitin Hydrolase BRCA1-Associated Protein 1 with Cell Cycle Regulator Host Cell Factor 1. Mol. Cell. Biol. 2009, 29, 2181–2192, doi:10.1128/mcb.01517-08.
- Forbes, S.A.; Bindal, N.; Bamford, S.; Cole, C.; Kok, C.Y.; Beare, D.; Jia, M.; Shepherd, R.; Leung, K.; Menzies, A.; et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2010, 39, D945–D950, doi:10.1093/nar/gkq929.
- Harris, S.L.; Levine, A.J. The p53 pathway: Positive and negative feedback loops. Oncogene 2005, 24, 2899–2908, doi:10.1038/sj.onc.1208615.
- Zhang, X.; Berger, F.G.; Yang, J.; Lu, X. USP4 inhibits p53 through deubiquitinating and stabilizing ARF-BP1. EMBO J. 2011, 30, 2177–2189, doi:10.1038/emboj.2011.125.
- Kruse, J.-P.; Gu, W. Modes of p53 regulation. Cell 2009, 137, 609–622, doi:10.1016/j.cell.2009.04.050.
- Kwon, S.-K.; Saindane, M.; Baek, K.-H. p53 stability is regulated by diverse deubiquitinating enzymes. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 404–411, doi:10.1016/j.bbcan.2017.08.001.
- Benassi, B.; Marani, M.; Loda, M.; Blandino, G. USP2a alters chemotherapeutic response by modulating redox. Cell Death Dis. 2013, 4, e812, doi:10.1038/cddis.2013.289.
- Kim, J.; Kim, W.-J.; Liu, Z.; Loda, M.F.; Freeman, M.R. The ubiquitin-specific protease USP2a enhances tumor progression by targeting cyclin A1 in bladder cancer. Cell Cycle 2012, 11, 1123–1130, doi:10.4161/cc.11.6.19550.
- Priolo, C.; Tang, D.; Brahamandan, M.; Benassi, B.; Sicinska, E.; Ogino, S.; Farsetti, A.; Porrello, A.; Finn, S.P.; Zimmermann, J.; et al. The Isopeptidase USP2a Protects Human Prostate Cancer from Apoptosis. Cancer Res. 2006, 66, 8625–8632, doi:10.1158/0008-5472.can-06-1374.
- Graner, E.; Tang, D.; Rossi, S.; Baron, A.; Migita, T.; Weinstein, L.J.; Lechpammer, M.; Huesken, D.; Zimmermann, J.; Signoretti, S.; et al. The isopeptidase USP2a regulates the stability of fatty acid synthase in prostate cancer. Cancer Cell 2004, 5, 253–261, doi:10.1016/s1535-6108(04)00055-8.
- Stevenson, L.F.; Sparks, A.; Allende-Vega, N.; Xirodimas, D.P.; Lane, D.P.; Saville, M.K. The deubiquitinating enzyme USP2a regulates the p53 pathway by targeting Mdm2. EMBO J. 2007, 26, 976–986, doi:10.1038/sj.emboj.7601567.
- Allende-Vega, N.; Sparks, A.; Lane, D.P.; Saville, M.K. MdmX is a substrate for the deubiquitinating enzyme USP2a. Oncogene 2009, 29, 432–441, doi:10.1038/onc.2009.330.
- Liu, Z.; Zanata, S.; Kim, J.; A Peterson, M.; Di Vizio, L.; Chirieac, L.R.; Pyne, S.; Agostini, M.; Freeman, M.R.; Loda, M. The ubiquitin-specific protease USP2a prevents endocytosis-mediated EGFR degradation. Oncogene 2012, 32, 1660–1669, doi:10.1038/onc.2012.188.
- Oh, K.H.; Yang, S.W.; Park, J.M.; Seol, J.H.; Iemura, S.; Natsume, T.; Murata, S.; Tanaka, K.; Jeon, Y.J.; Chung, C.H. Control of AIF-mediated cell death by antagonistic functions of CHIP ubiquitin E3 ligase and USP2 deubiquitinating enzyme. Cell Death Differ. 2011, 18, 1326–1336, doi:10.1038/cdd.2011.3.
- Shan, J.; Zhao, W.; Gu, W. Suppression of Cancer Cell Growth by Promoting Cyclin D1 Degradation. Mol. Cell 2009, 36, 469–476, doi:10.1016/j.molcel.2009.10.018.
- Shi, Y.; Solomon, L.R.; Pereda-Lopez, A.; Giranda, V.L.; Luo, Y.; Johnson, E.F.; Shoemaker, A.R.; Leverson, J.; Liu, X. Ubiquitin-specific Cysteine Protease 2a (USP2a) Regulates the Stability of Aurora-A. J. Biol. Chem. 2011, 286, 38960–38968, doi:10.1074/jbc.M111.231498.
- Wang, C.-L.; Wang, J.-Y.; Liu, Z.-Y.; Ma, X.-M.; Wang, X.-W.; Jin, H.; Zhang, X.-P.; Fu, D.; Hou, L.-J.; Lu, Y.-C. Ubiquitin-specific protease 2a stabilizes MDM4 and facilitates the p53-mediated intrinsic apoptotic pathway in glioblastoma. Carcinogenesis 2014, 35, 1500–1509, doi:10.1093/carcin/bgu015.
- Dayal, S.; Sparks, A.; Jacob, J.; Allende-Vega, N.; Lane, D.P.; Saville, M.K. Suppression of the deubiquitinating enzyme USP5 causes the accumulation of unanchored polyubiquitin and the activation of p53. J. Biol. Chem. 2008, 284, 5030–5041, doi:10.1074/jbc.M805871200.
- Brooks, C.L.; Li, M.; Hu, M.; Shi, Y.; Gu, W. The p53–Mdm2–HAUSP complex is involved in p53 stabilization by HAUSP. Oncogene 2007, 26, 7262–7266, doi:10.1038/sj.onc.1210531.
- Cummins, J.M.; Vogelstein, B. HAUSP is required for p53 destabilization. Cell Cycle 2004, 3, 687–690, doi:10.4161/cc.3.6.924.
- Van Loosdregt, J.; Fleskens, V.; Fu, J.; Brenkman, A.B.; Bekker, C.P.; Pals, C.E.; Meerding, J.; Berkers, C.R.; Barbi, J.; Grone, A.; et al. Stabilization of the transcription factor Foxp3 by the deubiquitinase USP7 increases Treg-cell-suppressive capacity. Immunity 2013, 39, 259–271, doi:10.1016/j.immuni.2013.05.018.
- Van Der Horst, A.; De Vries-Smits, A.M.; Brenkman, A.B.; Van Triest, M.H.; Van Den Broek, N.; Colland, F.; Maurice, M.M.; Burgering, B.M. FOXO4 transcriptional activity is regulated by monoubiquitination and USP7/HAUSP. Nat. Cell Biol. 2006, 8, 1064–1073.
- Song, M.S.; Salmena, L.; Carracedo, A.; Egia, A.; Coco, F.L.; Teruya-Feldstein, J.; Pandolfi, P.P. The deubiquitinylation and localization of PTEN are regulated by a HAUSP–PML network. Nature 2008, 455, 813–817, doi:10.1038/nature07290.
- Lecona, E.; Rodriguez-Acebes, S.; Specks, J.; Lopez-Contreras, A.J.; Ruppen, I.; Murga, M.; Munoz, J.; Mendez, J.; Fernandez-Capetillo, O. USP7 is a SUMO deubiquitinase essential for DNA replication. Nat. Struct. Mol. Biol. 2016, 23, 270–277, doi:10.1038/nsmb.3185.
- Yuan, J.; Luo, K.; Zhang, L.; Cheville, J.C.; Lou, Z. USP10 Regulates p53 Localization and Stability by Deubiquitinating p53. Cell 2010, 140, 384–396, doi:10.1016/j.cell.2009.12.032.
- Wu, Y.; Wang, Y.; Yang, X.H.; Kang, T.; Zhao, Y.; Wang, C.; Evers, B.M.; Zhou, B.P. The Deubiquitinase USP28 Stabilizes LSD1 and Confers Stem-Cell-like Traits to Breast Cancer Cells. Cell Rep. 2013, 5, 224–236, doi:10.1016/j.celrep.2013.08.030.
- Zhang, L.; Xu, B.; Qiang, Y.; Huang, H.; Wang, C.; Li, D.; Qian, J. Overexpression of deubiquitinating enzyme USP28 promoted non-small cell lung cancer growth. J. Cell. Mol. Med. 2015, 19, 799–805, doi:10.1111/jcmm.12426.
- Zhao, L.-J.; Zhang, T.; Feng, X.-J.; Chang, J.; Suo, F.-Z.; Ma, J.-L.; Liu, Y.-J.; Zheng, Y.-C.; Liu, H.-M. USP28 contributes to the proliferation and metastasis of gastric cancer. J. Cell. Biochem. 2018, 120, 7657–7666, doi:10.1002/jcb.28040.
- Guo, G.; Xu, Y.; Gong, M.; Cao, Y.; An, R. USP28 is a potential prognostic marker for bladder cancer. Tumor Biol. 2013, 35, 4017–4022, doi:10.1007/s13277-013-1525-1.
- Diefenbacher, M.E.; Popov, N.; Blake, S.M.; Schülein-Völk, C.; Nye, E.; Spencer-Dene, B.; Jaenicke, L.A.; Eilers, M.; Behrens, A. The deubiquitinase USP28 controls intestinal homeostasis and promotes colorectal cancer. J. Clin. Investig. 2014, 124, 3407–3418, doi:10.1172/JCI73733.
- Popov, N.; Wanzel, M.; Madiredjo, M.; Zhang, D.; Beijersbergen, R.; Bernards, R.; Moll, R.; Elledge, S.J.; Eilers, M. The ubiquitin-specific protease USP28 is required for MYC stability. Nature 2007, 9, 765–774, doi:10.1038/ncb1601.
- Cuella-Martin, R.; Oliveira, C.; Lockstone, H.E.; Snellenberg, S.; Grolmusova, N.; Chapman, J.R. 53BP1 Integrates DNA Repair and p53-Dependent Cell Fate Decisions via Distinct Mechanisms. Mol. Cell 2016, 64, 51–64, doi:10.1016/j.molcel.2016.08.002.
- Liu, J.; Chung, H.-J.; Vogt, M.; Jin, Y.; Malide, D.; He, L.; Dundr, M.; Levens, D. JTV1 co-activates FBP to induce USP29 transcription and stabilize p53 in response to oxidative stress. EMBO J. 2011, 30, 846–858, doi:10.1038/emboj.2011.11.
- Hock, A.K.; Vigneron, A.M.; Carter, S.; Ludwig, R.L.; Vousden, K.H. Regulation of p53 stability and function by the deubiquitinating enzyme USP42. EMBO J. 2011, 30, 4921–4930, doi:10.1038/emboj.2011.419.
- Hock, A.K.; Vigneron, A.M.; Vousden, K.H. Ubiquitin-specific peptidase 42 (USP42) functions to deubiquitylate histones and regulate transcriptional activity. J. Biol. Chem. 2014, 289, 34862–34870, doi:10.1074/jbc.M114.589267.
- Liu, H.; Chen, W.; Liang, C.; Chen, B.W.; Zhi, X.; Zhang, S.; Zheng, X.; Bai, X.; Liang, T.-B. WP1130 increases doxorubicin sensitivity in hepatocellular carcinoma cells through usp9x-dependent p53 degradation. Cancer Lett. 2015, 361, 218–225, doi:10.1016/j.canlet.2015.03.001.
- Zhang, L.; Gong, F. Involvement of USP24 in the DNA damage response. Mol. Cell. Oncol. 2015, 3, e1011888, doi:10.1080/23723556.2015.1011888.
- Liu, W.-T.; Huang, K.-Y.; Lu, M.-C.; Huang, H.-L.; Chen, C.-Y.; Cheng, Y.-L.; Yu, H.-C.; Liu, S.-Q.; Lai, N.-S.; Huang, H.-B. TGF-β upregulates the translation of USP15 via the PI3K/AKT pathway to promote p53 stability. Oncogene 2016, 36, 2715–2723, doi:10.1038/onc.2016.424.
- Ke, J.-Y.; Dai, C.-J.; Wu, W.-L.; Gao, J.-H.; Xia, A.-J.; Liu, G.-P.; Lv, K.-S.; Wu, C.-L. USP11 regulates p53 stability by deubiquitinating p53. J. Zhejiang Univ. Sci. B 2014, 15, 1032–1038, doi:10.1631/jzus.B1400180.
- Tu, R.; Kang, W.; Yang, X.; Zhang, Q.; Xie, X.; Liu, W.; Zhang, J.; Zhang, X.-D.; Wang, H.; Du, R.-L. USP49 participates in the DNA damage response by forming a positive feedback loop with p53. Cell Death Dis. 2018, 9, 553, doi:10.1038/s41419-018-0475-3.
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86, doi:10.1186/1476-4598-12-86.
- Harhaj, E.W.; Dixit, V.M. Regulation of NF-κB by deubiquitinases. Immunol. Rev. 2012, 246, 107–124, doi:10.1111/j.1600-065X.2012.01100.x.
- Hayden, M.S.; Ghosh, S. Shared principles in NF-κB signaling. Cell 2008, 132, 344–362.
- Massoumi, R.; Chmielarska, K.; Hennecke, K.; Pfeifer, A.; Fässler, R. Cyld inhibits tumor cell proliferation by blocking Bcl-3-dependent NF-κB signaling. Cell 2006, 125, 665–677.
- Zhang, J.; Stirling, B.; Temmerman, S.T.; Ma, C.A.; Fuss, I.J.; Derry, J.M.; Jain, A. Impaired regulation of NF-kappaB and increased susceptibility to colitis-associated tumorigenesis in CYLD-deficient mice. J. Clin. Invest. 2006, 116, 3042–3049, doi:10.1172/jci28746.
- Evans, P.C.; Ovaa, H.; Hamon, M.; Kilshaw, P.J.; Hamm, S.; Bauer, S.; Ploegh, H.L.; Smith, T.S. Zinc-finger protein A20, a regulator of inflammation and cell survival, has de-ubiquitinating activity. Biochem. J. 2004, 378, 727–734, doi:10.1042/bj20031377.
- Opipari, A.W.; Boguski, M.S.; Dixit, V.M. The A20 cDNA induced by tumor necrosis factor alpha encodes a novel type of zinc finger protein. J. Biol. Chem. 1990, 265, 14705–14708.
- Wertz, I.E.; O’Rourke, K.M.; Zhou, H.; Eby, M.; Aravind, L.; Seshagiri, S.; Wu, P.; Wiesmann, C.; Baker, R.; Boone, D.L.; et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature 2004, 430, 694–699, doi:10.1038/nature02794.
- Li, L.-Y.; Soetandyo, N.; Wang, Q.; Ye, Y. The zinc finger protein A20 targets TRAF2 to the lysosomes for degradation. Biochim. Biophys. Acta Bioenerg. 2009, 1793, 346–353, doi:10.1016/j.bbamcr.2008.09.013.
- Xu, G.; Tan, X.; Wang, H.; Sun, W.; Shi, Y.; Burlingame, S.; Gu, X.; Cao, G.; Zhang, T.; Qin, J.; et al. Ubiquitin-specific Peptidase 21 Inhibits Tumor Necrosis Factor α-induced Nuclear Factor κB Activation via Binding to and Deubiquitinating Receptor-interacting Protein 1. J. Biol. Chem. 2009, 285, 969–978, doi:10.1074/jbc.M109.042689.
- Hou, X.; Wang, L.; Zhang, L.; Pan, X.; Zhao, W. Ubiquitin-specific protease 4 promotes TNF-α-induced apoptosis by deubiquitination of RIP1 in head and neck squamous cell carcinoma. FEBS Lett. 2013, 587, 311–316, doi:10.1016/j.febslet.2012.12.016.
- Enesa, K.; Zakkar, M.; Chaudhury, H.; Luong, L.A.; Rawlinson, L.; Mason, J.C.; Haskard, D.O.; Dean, J.L.; Evans, P.C. NF-κB suppression by the deubiquitinating enzyme cezanne a novel negative feedback loop in pro-inflammatory signaling. J. Biol. Chem. 2008, 283, 7036–7045.
- McNally, R.S.; Davis, B.K.; Clements, C.; Accavitti-Loper, M.A.; Mak, T.W.; Ting, J.P.-Y. DJ-1 Enhances Cell Survival through the Binding of Cezanne, a Negative Regulator of NF-κB. J. Biol. Chem. 2010, 286, 4098–4106, doi:10.1074/jbc.M110.147371.
- Tzimas, C.; Michailidou, G.; Arsenakis, M.; Kieff, E.; Mosialos, G.; Hatzivassiliou, E.G. Human ubiquitin specific protease 31 is a deubiquitinating enzyme implicated in activation of nuclear factor-κB. Cell. Signal. 2006, 18, 83–92, doi:10.1016/j.cellsig.2005.03.017.
- Daubeuf, S.; Singh, D.; Tan, Y.; Liu, H.; Federoff, H.J.; Bowers, W.J.; Tolba, K. HSV ICP0 recruits USP7 to modulate TLR-mediated innate response. Blood 2009, 113, 3264–3275, doi:10.1182/blood-2008-07-168203.
- Metzig, M.; Nickles, D.; Falschlehner, C.; Lehmann-Koch, J.; Straub, B.; Roth, W.; Boutros, M. An RNAi screen identifies USP2 as a factor required for TNF-α-induced NF-κB signaling. Int. J. Cancer 2011, 129, 607–618, doi:10.1002/ijc.26124.
- González-Navajas, J.; Law, J.; Nguyen, K.P.; Bhargava, M.; Corr, M.P.; Varki, N.; Eckmann, L.; Hoffman, H.M.; Lee, J.; Raz, E. Interleukin 1 receptor signaling regulates DUBA expression and facilitates Toll-like receptor 9–driven antiinflammatory cytokine production. J. Exp. Med. 2010, 207, 2799–2807, doi:10.1084/jem.20101326.
- Schweitzer, K.; Bozko, P.M.; Dubiel, W.; Naumann, M. CSN controls NF-κB by deubiquitinylation of IκBα. EMBO J. 2007, 26, 1532–1541, doi:10.1038/sj.emboj.7601600.
- Yamaguchi, T.; Kimura, J.; Miki, Y.; Yoshida, K. The deubiquitinating enzyme USP11 controls an IκB kinase α (IKKα)-p53 signaling pathway in response to tumor necrosis factor α (TNFα). J. Biol. Chem. 2007, 282, 33943–33948.
- Takeuchi, K.; Ito, F. Receptor tyrosine kinases and targeted cancer therapeutics. Biol. Pharm. Bull. 2011, 34, 1774–1780, doi:10.1248/bpb.34.1774.
- Critchley, W.; Pellet-Many, C.; Ringham-Terry, B.; Harrison, M.; Zachary, I.C.; Ponnambalam, S. Receptor Tyrosine Kinase Ubiquitination and De-Ubiquitination in Signal Transduction and Receptor Trafficking. Cells 2018, 7, 22, doi:10.3390/cells7030022.
- Niendorf, S.; Oksche, A.; Kisser, A.; Löhler, J.; Prinz, M.; Schorle, H.; Feller, S.; Lewitzky, M.; Horak, I.; Knobeloch, K.-P. Essential Role of Ubiquitin-Specific Protease 8 for Receptor Tyrosine Kinase Stability and Endocytic Trafficking In Vivo. Mol. Cell. Biol. 2007, 27, 5029–5039, doi:10.1128/mcb.01566-06.
- Alwan, H.A.J.; Van Leeuwen, J.E.M. UBPY-mediated Epidermal Growth Factor Receptor (EGFR) De-ubiquitination Promotes EGFR Degradation. J. Biol. Chem. 2006, 282, 1658–1669, doi:10.1074/jbc.m604711200.
- Duex, J.E.; Sorkin, A. RNA Interference Screen Identifies Usp18 as a Regulator of Epidermal Growth Factor Receptor Synthesis. Mol. Biol. Cell 2009, 20, 1833–1844, doi:10.1091/mbc.E08-08-0880.
- Liu, H.; Buus, R.; Clague, M.J.; Urbe, S. Regulation of ErbB2 Receptor Status by the Proteasomal DUB POH1. PLoS ONE 2009, 4, e5544, doi:10.1371/journal.pone.0005544.
- Duex, J.E.; Comeau, L.; Sorkin, A.; Purow, B.; Kefas, B. Usp18 Regulates Epidermal Growth Factor (EGF) Receptor Expression and Cancer Cell Survival via MicroRNA-7. J. Biol. Chem. 2011, 286, 25377–25386, doi:10.1074/jbc.M111.222760.
- Savio, M.G.; Wollscheid, N.; Cavallaro, E.; Algisi, V.; Di Fiore, P.P.; Sigismund, S.; Maspero, E.; Polo, S. USP9X Controls EGFR Fate by Deubiquitinating the Endocytic Adaptor Eps15. Curr. Biol. 2016, 26, 173–183, doi:10.1016/j.cub.2015.11.050
- Pareja, F.; Ferraro, D.A.; Rubin, C.; Cohen-Dvashi, H.; Zhang, F.; Aulmann, S.; Ben-Chetrit, N.; Pines, G.; Navon, R.; Crosetto, N.; et al. Deubiquitination of EGFR by Cezanne-1 contributes to cancer progression. Oncogene 2011, 31, 4599–608, doi:10.1038/onc.2011.587.
- Klaus, A.; Birchmeier, W.; Klaus-Bergmann, A. Wnt signalling and its impact on development and cancer. Nat. Rev. Cancer 2008, 8, 387–398, doi:10.1038/nrc2389.
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26.
- Saito-Diaz, K.; Chen, T.W.; Wang, X.; Thorne, C.A.; Wallace, H.A.; Page-McCaw, A.; Lee, E. The way Wnt works: Components and mechanism. Growth Factors 2012, 31, 1–31, doi:10.3109/08977194.2012.752737.
- Katoh, M. WNT/PCP signaling pathway and human cancer. Oncol. Rep. 2005, 14, 1583–1588, doi:10.3892/or.14.6.1583.
- Luga, V.; Zhang, L.; Viloria-Petit, A.M.; Ogunjimi, A.A.; Inanlou, M.R.; Chiu, E.; Buchanan, M.; Hosein, A.N.; Basik, M.; Wrana, J.L. Exosomes Mediate Stromal Mobilization of Autocrine Wnt-PCP Signaling in Breast Cancer Cell Migration. Cell 2012, 151, 1542–1556, doi:10.1016/j.cell.2012.11.024.
- Wang, Y. Wnt/Planar cell polarity signaling: A new paradigm for cancer therapy. Mol. Cancer Ther. 2009, 8, 2103–2109, doi:10.1158/1535-7163.mct-09-0282.
- Marikawa, Y.; Elinson, R.P. β-TrCP is a negative regulator of the Wnt/β-catenin signaling pathway and dorsal axis formation in Xenopus embryos. Mech. Dev. 1998, 77, 75–80, doi:10.1016/s0925-4773(98)00134-8.
- Fei, C.; Li, Z.; Li, C.; Chen, Y.; Chen, Z.; He, X.; Mao, L.; Wang, X.; Zeng, R.; Li, L. Smurf1-Mediated Lys29-Linked Nonproteolytic Polyubiquitination of Axin Negatively Regulates Wnt/β-Catenin Signaling. Mol. Cell. Biol. 2013, 33, 4095–4105, doi:10.1128/MCB.00418-13.
- Nielsen, C.; Jernigan, K.K.; Diggins, N.L.; Webb, D.J.; MacGurn, J.A. USP9X Deubiquitylates DVL2 to Regulate WNT Pathway Specification. Cell Rep. 2019, 28, 1074–1089.e5, doi:10.1016/j.celrep.2019.06.083.
- Jung, H.; Kim, B.-G.; Han, W.H.; Lee, J.H.; Cho, J.-Y.; Park, W.S.; Maurice, M.M.; Han, J.-K.; Lee, M.J.; Finley, D.; et al. Deubiquitination of Dishevelled by Usp14 is required for Wnt signaling. Oncogenesis 2013, 2, e64, doi:10.1038/oncsis.2013.28.
- Madan, B.; Walker, M.P.; Young, R.; Quick, L.; Orgel, K.A.; Ryan, M.; Gupta, P.; Henrich, I.C.; Ferrer, M.; Marine, S.; et al. USP6 oncogene promotes Wnt signaling by deubiquitylating Frizzleds. Proc. Natl. Acad. Sci. USA 2016, 113, E2945–E2954, doi:10.1073/pnas.1605691113.
- Yun, S.-I.; Kim, H.H.; Yoon, J.H.; Park, W.S.; Hahn, M.-J.; Kim, H.C.; Chung, C.H.; Kim, K.K. Ubiquitin specific protease 4 positively regulates the WNT/β-catenin signaling in colorectal cancer. Mol. Oncol. 2015, 9, 1834–1851, doi:10.1016/j.molonc.2015.06.006.
- Lui, T.T.H.; Lacroix, C.; Ahmed, S.M.; Goldenberg, S.J.; Leach, C.A.; Daulat, A.M.; Angers, S. The Ubiquitin-Specific Protease USP34 Regulates Axin Stability and Wnt/β-Catenin Signaling. Mol. Cell. Biol. 2011, 31, 2053–2065, doi:10.1128/MCB.01094-10.
- Pardali, K.; Moustakas, A. Actions of TGF-β as tumor suppressor and pro-metastatic factor in human cancer. Biochim. Biophys. Acta Bioenerg. 2007, 1775, 21–62, doi:10.1016/j.bbcan.2006.06.004.
- Dupont, S.; Mamidi, A.; Cordenonsi, M.; Montagner, M.; Zacchigna, L.; Adorno, M.; Martello, G.; Stinchfield, M.J.; Soligo, S.; Morsut, L.; et al. FAM/USP9x, a Deubiquitinating Enzyme Essential for TGFβ Signaling, Controls Smad4 Monoubiquitination. Cell 2009, 136, 123–135, doi:10.1016/j.cell.2008.10.051.
- Al-Hakim, A.K.; Zagorska, A.; Chapman, L.; Deak, M.; Peggie, M.; Alessi, D.R. Control of AMPK-related kinases by USP9X and atypical Lys29/Lys33-linked polyubiquitin chains. Biochem. J. 2008, 411, 249–260.
- Inui, M.; Manfrin, A.; Mamidi, A.; Martello, G.; Morsut, L.; Soligo, S.; Enzo, E.; Moro, S.; Polo, S.; Dupont, S.; et al. USP15 is a deubiquitylating enzyme for receptor-activated SMADs. Nature 2011, 13, 1368–1375, doi:10.1038/ncb2346.
- Ibarrola, N.; Kratchmarova, I.; Nakajima, D.; Schiemann, W.P.; Moustakas, A.; Pandey, A.; Mann, M. Cloning of a novel signaling molecule, AMSH-2, that potentiates transforming growth factor β signaling. BMC Cell Biol. 2004, 5, 2, doi:10.1186/1471-2121-5-2.
- Wicks, S.J.; Haros, K.; Maillard, M.; Song, L.; Cohen, R.E.; ten Dijke, P.; Chantry, A. The deubiquitinating enzyme UCH37 interacts with Smads and regulates TGF-β signalling. Oncogene 2005, 24, 8080–8084.
- Holland, A.J.; Cleveland, D.W. The deubiquitinase USP44 is a tumor suppressor that protects against chromosome missegregation. J. Clin. Investig. 2012, 122, 4325–4328, doi:10.1172/JCI66420.
- Huang, X.; Summers, M.K.; Pham, V.; Lill, J.R.; Liu, J.; Lee, G.; Kirkpatrick, D.S.; Jackson, P.K.; Fang, G.; Dixit, V.M. Deubiquitinase USP37 Is Activated by CDK2 to Antagonize APCCDH1 and Promote S Phase Entry. Mol. Cell 2011, 42, 511–523, doi:10.1016/j.molcel.2011.03.027.
- Faronato, M.; Patel, V.; Darling, S.; Dearden, L.; Clague, M.J.; Urbe, S.; Coulson, J.M. The deubiquitylase USP15 stabilizes newly synthesized REST and rescues its expression at mitotic exit. Cell Cycle 2013, 12, 1964–1977, doi:10.4161/cc.25035.
- McFarlane, C.; Kelvin, A.A.; De La Vega, M.; Govender, U.; Scott, C.J.; Burrows, J.F.; Johnston, J.A. The Deubiquitinating Enzyme USP17 Is Highly Expressed in Tumor Biopsies, Is Cell Cycle Regulated, and Is Required for G1-S Progression. Cancer Res. 2010, 70, 3329–3339, doi:10.1158/0008-5472.can-09-4152.
- Van Leuken, R.J.; Luna-Vargas, M.P.; Sixma, T.K.; Wolthuis, R.M.; Medema, R. Usp39 is essential for mitotic spindle checkpoint integrity and controls mRNA-levels of Aurora B. Cell Cycle 2008, 7, 2710–2719, doi:10.4161/cc.7.17.6553.
- Stegmeier, F.; Sowa, M.E.; Nalepa, G.; Gygi, S.P.; Harper, J.W.; Elledge, S.J. The tumor suppressor CYLD regulates entry into mitosis. Proc. Natl. Acad. Sci. USA 2007, 104, 8869–8874, doi:10.1073/pnas.0703268104.
- Nicassio, F.; Corrado, N.; Vissers, J.H.; Areces, L.B.; Bergink, S.; Marteijn, J.A.; Geverts, B.; Houtsmuller, A.C.; Vermeulen, W.; Di Fiore, P.P.; et al. Human USP3 is a chromatin modifier required for s phase progression and genome stability. Curr. Biol. 2007, 17, 1972–1977, doi:10.1016/j.cub.2007.10.034.
- Atanassov, B.; Koutelou, E.; Dent, S.Y. The role of deubiquitinating enzymes in chromatin regulation. FEBS Lett. 2010, 585, 2016–2023, doi:10.1016/j.febslet.2010.10.042.
- Cao, J.; Yan, Q. Histone ubiquitination and deubiquitination in transcription, DNA damage response, and cancer. Front Oncol. 2012, 2, 26, doi:10.3389/fonc.2012.00026.
- Joo, H.-Y.; Jones, A.; Yang, C.; Zhai, L.; Smith, A.D.; Zhang, Z.; Chandrasekharan, M.B.; Sun, Z.-W.; Renfrow, M.B.; Wang, Y.; et al. Regulation of Histone H2A and H2B Deubiquitination and Xenopus Development by USP12 and USP46. J. Biol. Chem. 2010, 286, 7190–7201, doi:10.1074/jbc.M110.158311.
- Zhang, X.-Y.; Pfeiffer, H.K.; Thorne, A.W.; McMahon, S.B. USP22, an hSAGA subunit and potential cancer stem cell marker, reverses the polycomb-catalyzed ubiquitylation of histone H2A. Cell Cycle 2008, 7, 1522–1524, doi:10.4161/cc.7.11.5962.
- Zhang, X.-Y.; Varthi, M.; Sykes, S.M.; Phillips, C.; Warzecha, C.; Zhu, W.; Wyce, A.; Thorne, A.W.; Berger, S.L.; McMahon, S.B. The Putative Cancer Stem Cell Marker USP22 Is a Subunit of the Human SAGA Complex Required for Activated Transcription and Cell-Cycle Progression. Mol. Cell 2008, 29, 102–111, doi:10.1016/j.molcel.2007.12.015.
- Draker, R.; Sarcinella, E.; Cheung, P. USP10 deubiquitylates the histone variant H2A.Z and both are required for androgen receptor-mediated gene activation. Nucleic Acids Res. 2011, 39, 3529–3542, doi:10.1093/nar/gkq1352.
- Maertens, G.; El Messaoudi-Aubert, S.; Elderkin, S.; Hiom, K.; Peters, G. Ubiquitin-specific proteases 7 and 11 modulate Polycomb regulation of the INK4a tumour suppressor. EMBO J. 2010, 29, 2553–2565, doi:10.1038/emboj.2010.129.
- Sowa, M.E.; Bennett, E.J.; Gygi, S.P.; Harper, J.W. Defining the Human Deubiquitinating Enzyme Interaction Landscape. Cell 2009, 138, 389–403, doi:10.1016/j.cell.2009.04.042.
- Song, E.J.; Werner, S.L.; Neubauer, J.; Stegmeier, F.; Aspden, J.L.; Rio, D.; Harper, J.W.; Elledge, S.J.; Kirschner, M.W.; Rape, M. The Prp19 complex and the Usp4Sart3 deubiquitinating enzyme control reversible ubiquitination at the spliceosome. Genes Dev. 2010, 24, 1434–1447, doi:10.1101/gad.1925010.
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078, doi:10.1038/nature08467.
- Huang, T.T.; Nijman, S.M.; Mirchandani, K.D.; Galardy, P.J.; Cohn, M.A.; Haas, W.; Gygi, S.P.; Ploegh, H.L.; Bernards, R.; D’Andrea, A.D. Regulation of monoubiquitinated PCNA by DUB autocleavage. Nature 2006, 8, 341–347, doi:10.1038/ncb1378.
- Nijman, S.M.; Huang, T.T.; Dirac, A.M.; Brummelkamp, T.R.; Kerkhoven, R.M.; D’Andrea, A.; Bernards, R. The Deubiquitinating Enzyme USP1 Regulates the Fanconi Anemia Pathway. Mol. Cell 2005, 17, 331–339, doi:10.1016/j.molcel.2005.01.008.
- Guervilly, J.-H.; Renaud, E.; Takata, M.; Rosselli, F. USP1 deubiquitinase maintains phosphorylated CHK1 by limiting its DDB1-dependent degradation. Hum. Mol. Genet. 2011, 20, 2171–2181, doi:10.1093/hmg/ddr103.
- Williams, S.; Maecker, H.L.; French, R.M.; Liu, J.; Gregg, A.; Silverstein, L.B.; Cao, T.C.; Carano, R.A.; Dixit, V.M. USP1 Deubiquitinates ID Proteins to Preserve a Mesenchymal Stem Cell Program in Osteosarcoma. Cell 2011, 146, 918–930, doi:10.1016/j.cell.2011.07.040.
- Matsuoka, S.; A Ballif, B.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage. Science 2007, 316, 1160–1166, doi:10.1126/science.1140321.
- Mu, J.-J.; Wang, Y.; Luo, H.; Leng, M.; Zhang, J.; Yang, T.; Besusso, D.; Jung, S.Y.; Qin, J. A proteomic analysis of ataxia telangiectasia-mutated (ATM)/ATM-Rad3-related (ATR) substrates identifies the ubiquitin-proteasome system as a regulator for DNA damage checkpoints. J. Biol. Chem. 2007, 282, 17330–17334.
- Zhang, N.; Zaugg, K.; Mak, T.W.; Elledge, S.J. A Role for the Deubiquitinating Enzyme USP28 in Control of the DNA-Damage Response. Cell 2006, 126, 529–542, doi:10.1016/j.cell.2006.06.039.
- Al-Hakim, A.; Escribano-Diaz, C.; Landry, M.-C.; O’Donnell, L.; Panier, S.; Szilard, R.K.; Durocher, D. The ubiquitous role of ubiquitin in the DNA damage response. DNA Repair 2010, 9, 1229–1240, doi:10.1016/j.dnarep.2010.09.011.
- McGarry, E.; Gaboriau, D.C.A.; Rainey, M.D.; Restuccia, U.; Bachi, A.; Santocanale, C. The deubiquitinase USP9X maintains DNA replication fork stability and DNA damage checkpoint responses by regulating CLASPIN during S-phase. Cancer Res. 2016, 76, 2384–2393, doi:10.1158/0008-5472.can-15-2890.
- Wolfsperger, F.; A Hogh-Binder, S.; Schittenhelm, J.; Psaras, T.; Ritter, V.; Bornes, L.; Huber, S.M.; Jendrossek, V.; Rudner, J. Deubiquitylating enzyme USP9x regulates radiosensitivity in glioblastoma cells by Mcl-1-dependent and -independent mechanisms. Cell Death Dis. 2016, 7, e2039, doi:10.1038/cddis.2015.405.
- Tian, Z.; D’Arcy, P.; Wang, X.; Ray, A.; Tai, Y.-T.; Hu, Y.; Carrasco, R.D.; Richardson, P.; Linder, S.; Chauhan, D.; et al. A novel small molecule inhibitor of deubiquitylating enzyme USP14 and UCHL5 induces apoptosis in multiple myeloma and overcomes bortezomib resistance. Blood 2014, 123, 706–716, doi:10.1182/blood-2013-05-500033.
- Fukui, S.; Nagasaka, K.; Miyagawa, Y.; Kikuchi-Koike, R.; Kawata, Y.; Kanda, R.; Ichinose, T.; Sugihara, T.; Hiraike, H.; Wada-Hiraike, O.; et al. The proteasome deubiquitinase inhibitor bAP15 downregulates TGF-β/Smad signaling and induces apoptosis via UCHL5 inhibition in ovarian cancer. Oncotarget 2019, 10, 5932–5948, doi:10.18632/oncotarget.27219.
- Wang, X.; Mazurkiewicz, M.; Hillert, E.-K.; Olofsson, M.H.; Pierrou, S.; Hillertz, P.; Gullbo, J.; Selvaraju, K.; Paulus, A.; Akhtar, S.; et al. The proteasome deubiquitinase inhibitor VLX1570 shows selectivity for ubiquitin-specific protease-14 and induces apoptosis of multiple myeloma cells. Sci. Rep. 2016, 6, 1–15.
- Ward, J.A.; Pinto-Fernandez, A.; Cornelissen, L.; Bonham, S.; Díaz-Sáez, L.; Riant, O.; Huber, K.V.M.; Kessler, B.M.; Feron, O.; Tate, E.W. Re-Evaluating the Mechanism of Action of α,β-Unsaturated Carbonyl DUB Inhibitors b-AP15 and VLX1570: A Paradigmatic Example of Unspecific Protein Cross-linking with Michael Acceptor Motif-Containing Drugs. J. Med. Chem. 2020, 63, 3756–3762, doi:10.1021/acs.jmedchem.0c00144.
- Rahmani, A.H.; Alsahli, M.A.; Aly, S.M.; Khan, M.A.; Aldebasi, Y.H. Role of Curcumin in Disease Prevention and Treatment. Adv. Biomed. Res. 2018, 7, 38, doi:10.4103/abr.abr_147_16.
- Jana, N.R.; Dikshit, P.; Goswami, A.; Nukina, N. Inhibition of Proteasomal Function by Curcumin Induces Apoptosis through Mitochondrial Pathway. J. Biol. Chem. 2003, 279, 11680–11685, doi:10.1074/jbc.m310369200.
- Zhou, B.; Zuo, Y.; Li, B.; Wang, H.; Liu, H.; Wang, X.; Qiu, X.; Hu, Y.; Wen, S.; Du, J.; et al. Deubiquitinase Inhibition of 19S Regulatory Particles by 4-Arylidene Curcumin Analog AC17 Causes NF- B Inhibition and p53 Reactivation in Human Lung Cancer Cells. Mol. Cancer Ther. 2013, 12, 1381–1392, doi:10.1158/1535-7163.mct-12-1057.
- Li, Z.; Melandri, F.; Berdo, I.; Jansen, M.; Hunter, L.; Wright, S.; Valbrun, D.; E Figueiredo-Pereira, M. Δ12-Prostaglandin J2 inhibits the ubiquitin hydrolase UCH-L1 and elicits ubiquitin–protein aggregation without proteasome inhibition. Biochem. Biophys. Res. Commun. 2004, 319, 1171–1180, doi:10.1016/j.bbrc.2004.05.098.
- Kawakita, T.; Masato, N.; Takiguchi, E.; Abe, A.; Irahara, M. Cytotoxic effects of 15-deoxy-Δ12,14-prostaglandin J2 alone and in combination with dasatinib against uterine sarcoma in vitro. Exp. Ther. Med. 2017, 13, 2939–2945, doi:10.3892/etm.2017.4346.
- Bie, Q.; Dong, H.; Jin, C.; Zhang, H.; Zhang, B. 15d-PGJ2 is a new hope for controlling tumor growth. Am. J. Transl. Res. 2018, 10, 648–658.
- Liu, H.; Li, W.; Ahmad, M.; Miller, T.M.; Rose, M.E.; Poloyac, S.M.; Uechi, G.; Balasubramani, M.; Hickey, R.W.; Graham, S. Modification of ubiquitin-C-terminal hydrolase-L1 by cyclopentenone prostaglandins exacerbates hypoxic injury. Neurobiol. Dis. 2010, 41, 318–328, doi:10.1016/j.nbd.2010.09.020.
- Aleo, E.; Henderson, C.J.; Fontanini, A.; Solazzo, B.; Brancolini, C. Identification of New Compounds That Trigger Apoptosome-Independent Caspase Activation and Apoptosis. Cancer Res. 2006, 66, 9235–9244, doi:10.1158/0008-5472.can-06-0702.
- Nicholson, B.; Leach, C.A.; Goldenberg, S.J.; Francis, D.M.; Kodrasov, M.P.; Tian, X.; Shanks, J.; Sterner, D.E.; Bernal, A.; Mattern, M.R.; et al. Characterization of ubiquitin and ubiquitin-like-protein isopeptidase activities. Protein Sci. 2008, 17, 1035–1043, doi:10.1110/ps.083450408.
- Fontanini, A.; Foti, C.; Potu, H.; Crivellato, E.; Maestro, R.; Bernardi, P.; Demarchi, F.; Brancolini, C. The Isopeptidase Inhibitor G5 Triggers a Caspase-independent Necrotic Death in Cells Resistant to Apoptosis. J. Biol. Chem. 2009, 284, 8369–8381, doi:10.1074/jbc.M806113200.
- Issaenko, O.A.; Amerik, A.Y. Chalcone-based small-molecule inhibitors attenuate malignant phenotype via targeting deubiquitinating enzymes. Cell Cycle 2012, 11, 1804–1817, doi:10.4161/cc.20174.
- Coughlin, K.; Anchoori, R.; Iizuka, Y.; Meints, J.; MacNeill, L.; Vogel, R.I.; Orlowski, R.Z.; Lee, M.K.; Roden, R.B.; Bazzaro, M. Small-molecule RA-9 inhibits proteasome-associated DUBs and ovarian cancer in vitro and in vivo via exacerbating unfolded protein responses. Clin. Cancer Res. 2014, 20, 3174–3186, doi:10.1158/1078-0432.CCR-13-2658.
- Kapuria, V.; Peterson, L.F.; Fang, D.; Bornmann, W.G.; Talpaz, M.; Donato, N.J. Deubiquitinase Inhibition by Small-Molecule WP1130 Triggers Aggresome Formation and Tumor Cell Apoptosis. Cancer Res. 2010, 70, 9265–9276, doi:10.1158/0008-5472.can-10-1530.
- Xiang, C.; Bao, Y.; Dong, Z.; Chen, Q.; Guo, H.; Ziang, C.; Shao, J. WP1130 attenuates cisplatin resistance by decreasing P53 expression in non-small cell lung carcinomas. Oncotarget 2017, 8, 49033–49043, doi:10.18632/oncotarget.16931.
- Kapuria, V.; Levitzki, A.; Bornmann, W.G.; Maxwell, D.; Priebe, W.; Sorenson, R.J.; Showalter, H.D.; Talpaz, M.; Donato, N.J. A novel small molecule deubiquitinase inhibitor blocks Jak2 signaling through Jak2 ubiquitination. Cell. Signal. 2011, 23, 2076–2085, doi:10.1016/j.cellsig.2011.08.002.
- Ritorto, M.S.; Ewan, R.; Perez-Oliva, A.B.; Knebel, A.; Buhrlage, S.J.; Wightman, M.; Kelly, S.; Wood, N.T.; Virdee, S.; Gray, N.S.; et al. Screening of DUB activity and specificity by MALDI-TOF mass spectrometry. Nat. Commun. 2014, 5, 4763, doi:10.1038/ncomms5763.
- Hu, C.; Zhang, M.; Moses, N.; Hu, C.-L.; Polin, L.; Chen, W.; Jang, H.; Heyza, J.; Malysa, A.; Caruso, J.A.; et al. The USP10-HDAC6 axis confers cisplatin resistance in non-small cell lung cancer lacking wild-type p53. Cell Death Dis. 2020, 11, 328, doi:10.1038/s41419-020-2519-8.
- Weisberg, E.; Schauer, N.J.; Yang, J.; Lamberto, I.; Doherty, L.; Bhatt, S.; Nonami, A.; Meng, C.; Letai, A.; Wright, R.; et al. Inhibition of USP10 induces degradation of oncogenic FLT3. Nat. Methods 2017, 13, 1207–1215, doi:10.1038/nchembio.2486.
- Weinstock, J.; Wu, J.; Cao, P.; Kingsbury, W.D.; McDermott, J.L.; Kodrasov, M.P.; McKelvey, D.M.; Kumar, K.G.S.; Goldenberg, S.J.; Mattern, M.R.; et al. Selective Dual Inhibitors of the Cancer-Related Deubiquitylating Proteases USP7 and USP47. ACS Med. Chem. Lett. 2012, 3, 789–792, doi:10.1021/ml200276j.
- Colland, F.; Formstecher, E.; Jacq, X.; Reverdy, C.; Planquette, C.; Conrath, S.; Trouplin, V.; Bianchi, J.; Aushev, V.N.; Camonis, J. Small-molecule inhibitor of USP7/HAUSP ubiquitin protease stabilizes and activates p53 in cells. Mol. Cancer Ther. 2009, 8, 2286–2295.
- Reverdy, C.; Conrath, S.; Lopez, R.; Planquette, C.; Atmanene, C.; Collura, V.; Harpon, J.; Battaglia, V.; Vivat, V.; Sippl, W. Discovery of specific inhibitors of human USP7/HAUSP deubiquitinating enzyme. Chem. Biol. 2012, 19, 467–477.
- Cremer, A.; Stegmaier, K. Targeting DUBs to degrade oncogenic proteins. Br. J. Cancer 2020, 122, 1121–1123, doi:10.1038/s41416-020-0728-7.
- Pozhidaeva, A.; Valles, G.; Wang, F.; Wu, J.; Sterner, D.E.; Nguyen, P.; Weinstock, J.; Kumar, K.S.; Kanyo, J.; Wright, D.L.; et al. USP7-Specific Inhibitors Target and Modify the Enzyme’s Active Site via Distinct Chemical Mechanisms. Cell Chem. Biol. 2017, 24, 1501–1512, doi:10.1016/j.chembiol.2017.09.004.
- Chauhan, D.; Tian, Z.; Nicholson, B.; Kumar, K.S.; Zhou, B.; Carrasco, R.; McDermott, J.L.; Leach, C.A.; Fulcinniti, M.; Kodrasov, M.P.; et al. A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell 2012, 22, 345–358, doi:10.1016/j.ccr.2012.08.007.
- Schauer, N.J.; Liu, X.; Magin, R.S.; Doherty, L.M.; Chan, W.C.; Ficarro, S.B.; Hu, W.; Roberts, R.M.; Iacob, R.E.; Stolte, B.; et al. Selective USP7 inhibition elicits cancer cell killing through a p53-dependent mechanism. Sci. Rep. 2020, 10, 1–15, doi:10.1038/s41598-020-62076-x.
- Gavory, G.; O’Dowd, C.; McClelland, K.; Odrzywol, E.; Brown, A.; Burton, S.; Barker, O.; Burkamp, F.; Helm, M.; James, I.; et al. Abstract LB-257: Discovery and characterization of novel, highly potent and selective USP7 inhibitors. Exp. Mol. Ther. 2015, 75, doi:10.1158/1538-7445.am2015-lb-257.
- Kategaya, L.; Di Lello, P.; Rougé, L.; Pastor, R.; Clark, K.R.; Drummond, J.; Kleinheinz, T.; Lin, E.; Upton, J.-P.; Prakash, S.; et al. USP7 small-molecule inhibitors interfere with ubiquitin binding. Nature 2017, 550, 534–538, doi:10.1038/nature24006.
- Turnbull, A.P.; Ioannidis, S.; Krajewski, W.W.; Pinto-Fernandez, A.; Heride, C.; Martin, A.C.L.; Tonkin, L.M.; Townsend, E.C.; Buker, S.M.; Lancia, D.R.; et al. Molecular basis of USP7 inhibition by selective small-molecule inhibitors. Nature 2017, 550, 481–486, doi:10.1038/nature24451.
- Davis, M.I.; Pragani, R.; Fox, J.T.; Shen, M.; Parmar, K.; Gaudiano, E.F.; Liu, L.; Tanega, C.; McGee, L.; Hall, M.D.; et al. Small Molecule Inhibition of the Ubiquitin-specific Protease USP2 Accelerates cyclin D1 Degradation and Leads to Cell Cycle Arrest in Colorectal Cancer and Mantle Cell Lymphoma Models. J. Biol. Chem. 2016, 291, 24628–24640, doi:10.1074/jbc.M116.738567.
- Tomala, M.D.; Magiera-Mularz, K.; Kubica, K.; Krzanik, S.; Zięba, B.; Musielak, B.; Pustuła, M.; Popowicz, G.M.; Sattler, M.; Dubin, G.; et al. Identification of small-molecule inhibitors of USP2a. Eur. J. Med. Chem. 2018, 150, 261–267, doi:10.1016/j.ejmech.2018.03.009.
- Okada, K.; Ye, Y.Q.; Taniguchi, K.; Yoshida, A.; Akiyama, T.; Yoshioka, Y.; Onose, J.-I.; Koshino, H.; Takahashi, S.; Yajima, A.; et al. Vialinin A is a ubiquitin-specific peptidase inhibitor. Bioorganic Med. Chem. Lett. 2013, 23, 4328–4331, doi:10.1016/j.bmcl.2013.05.093.
- Sonowal, H.; Shukla, K.; Kota, S.; Saxena, A.; Ramana, K.V. Vialinin a, an edible mushroom-derived p-terphenyl antioxidant, prevents vegf-induced neovascularization in vitro and in vivo. Oxidative Med. Cell. Longev. 2018, 2018, 1–10, doi:10.1155/2018/1052102.
- Byun, S.; Lee, S.-Y.; Lee, J.; Jeong, C.-H.; Farrand, L.; Lim, S.; Reddy, K.; Kim, J.Y.; Lee, M.-H.; Lee, H.J.; et al. USP8 is a novel target for overcoming gefitinib resistance in lung cancer. Clin. Cancer Res. 2013, 19, 3894–3904, doi:10.1158/1078-0432.CCR-12-3696.
- Colombo, M.; Vallese, S.; Peretto, I.; Jacq, X.; Rain, J.-C.; Colland, F.; Guédat, P. Synthesis and Biological Evaluation of 9-Oxo-9H-indeno[1,2-b]pyrazine-2,3-dicarbonitrile Analogues as Potential Inhibitors of Deubiquitinating Enzymes. ChemMedChem 2010, 5, 552–558, doi:10.1002/cmdc.200900409.
- Lee, B.-H.; Lee, M.J.; Park, S.; Oh, N.-C.; Elsasser, S.; Chen, P.-C.; Gartner, C.; Dimova, N.; Hanna, J.; Gygi, S.P.; et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 2010, 467, 179–184, doi:10.1038/nature09299.
- Ma, Y.-S.; Wang, X.-F.; Zhang, Y.-J.; Luo, P.; Long, H.-D.; Li, L.; Yang, H.-Q.; Xie, R.-T.; Jia, C.-Y.; Lu, G.-X.; et al. Inhibition of USP14 Deubiquitinating Activity as a Potential Therapy for Tumors with p53 Deficiency. Mol. Ther. Oncolytics 2020, 16, 147–157, doi:10.1016/j.omto.2019.12.013.
- Liu, Z.; Zhao, T.; Li, Z.; Sun, K.; Fu, Y.; Cheng, T.; Guo, J.; Yu, B.; Shi, X.; Liu, H. Discovery of [1,2,3]triazolo[4,5-d]pyrimidine derivatives as highly potent, selective, and cellularly active USP28 inhibitors. Acta Pharm. Sin. B 2019, doi:10.1016/j.apsb.2019.12.008.
- Mermerian, A.H.; Case, A.; Stein, R.L.; Cuny, G.D. Structure–activity relationship, kinetic mechanism, and selectivity for a new class of ubiquitin C-terminal hydrolase-L1 (UCH-L1) inhibitors. Bioorganic. Med. Chem. Lett. 2007, 17, 3729–3732, doi:10.1016/j.bmcl.2007.04.027.
- Liu, Y.; Lashuel, H.A.; Choi, S.; Xing, X.; Case, A.; Ni, J.; Yeh, L.-A.; Cuny, G.D.; Stein, R.L.; Lansbury, P.T. Discovery of Inhibitors that Elucidate the Role of UCH-L1 Activity in the H1299 Lung Cancer Cell Line. Chem. Biol. 2003, 10, 837–846, doi:10.1016/j.chembiol.2003.08.010.
- Kobayashi, E.; Hwang, D.; Bheda-Malge, A.; Whitehurst, C.B.; Kabanov, A.V.; Kondo, S.; Aga, M.; Yoshizaki, T.; Pagano, J.S.; Sokolsky, M.; et al. Inhibition of UCH-L1 Deubiquitinating Activity with Two Forms of LDN-57444 Has Anti-Invasive Effects in Metastatic Carcinoma Cells. Int. J. Mol. Sci. 2019, 20, 3733, doi:10.3390/ijms20153733.
- Chen, J.; Dexheimer, T.S.; Ai, Y.; Liang, Q.; Villamil, M.A.; Inglese, J.; Maloney, D.J.; Jadhav, A.; Simeonov, A.; Zhuang, Z. Selective and Cell-Active Inhibitors of the USP1/ UAF1 Deubiquitinase Complex Reverse Cisplatin Resistance in Non-small Cell Lung Cancer Cells. Chem. Biol. 2011, 18, 1390–400, doi:10.1016/j.chembiol.2011.08.014.
- Liang, Q.; Dexheimer, T.S.; Zhang, P.; Rosenthal, A.S.; A Villamil, M.; You, C.; Zhang, Q.; Chen, J.; A Ott, C.; Sun, H.; et al. A selective USP1–UAF1 inhibitor links deubiquitination to DNA damage responses. Nat. Methods 2014, 10, 298–304, doi:10.1038/nchembio.1455.
- Dexheimer, T.S.; Rosenthal, A.S.; Luci, D.K.; Liang, Q.; Villamil, M.A.; Chen, J.; Sun, H.; Kerns, E.H.; Simeonov, A.; Jadhav, A.; et al. Synthesis and Structure–Activity Relationship Studies of N-Benzyl-2-phenylpyrimidin-4-amine Derivatives as Potent USP1/UAF1 Deubiquitinase Inhibitors with Anticancer Activity against Nonsmall Cell Lung Cancer. J. Med. Chem. 2014, 57, 8099–8110, doi:10.1021/jm5010495.
- A Burkhart, R.; Peng, Y.; Norris, Z.A.; Tholey, R.M.; Talbott, V.A.; Liang, Q.; Ai, Y.; Miller, K.; Lal, S.; A Cozzitorto, J.; et al. Mitoxantrone Targets Human Ubiquitin-Specific Peptidase 11 (USP11) and Is a Potent Inhibitor of Pancreatic Cancer Cell Survival. Mol. Cancer Res. 2013, 11, 901–911, doi:10.1158/1541-7786.MCR-12-0699.
- Wang, Q.; Li, L.-Y.; Ye, Y. Inhibition of p97-dependent protein degradation by Eeyarestatin I. J. Biol. Chem. 2008, 283, 7445–7454, doi:10.1074/jbc.M708347200.
- Biju, M.; Su, D.; Fisher, K.; Yang, J.; Robell, K.; Bobko, M. Discovery of potent and selective small molecule USP20 inhibitors. In Proceedings of the Ubiquitin Drug Discovery and Diagnostics Conference, Philadelphia, PA, USA, 13 October 2010.
- Altun, M.; Kramer, H.; Willems, L.I.; McDermott, J.L.; Leach, C.; Goldenberg, S.J.; Kumar, K.S.; Konietzny, R.; Fischer, R.; Kogan, E.; et al. Activity-Based Chemical Proteomics Accelerates Inhibitor Development for Deubiquitylating Enzymes. Chem. Biol. 2011, 18, 1401–1412, doi:10.1016/j.chembiol.2011.08.018.
- Reiner, T.; Parrondo, R.; Pozas, A.D.L.; Palenzuela, D.; Perez-Stable, C. Betulinic Acid Selectively Increases Protein Degradation and Enhances Prostate Cancer-Specific Apoptosis: Possible Role for Inhibition of Deubiquitinase Activity. PLoS ONE 2013, 8, e56234, doi:10.1371/journal.pone.0056234.
- Sanz, J.D.J.; Núñez, E.; Corcuera, B.L.; Aragon, C. Constitutive Endocytosis and Turnover of the Neuronal Glycine Transporter GlyT2 Is Dependent on Ubiquitination of a C-Terminal Lysine Cluster. PLoS ONE 2013, 8, e58863, doi:10.1371/journal.pone.0058863.
- Peterson, L.F.; Sun, H.; Liu, Y.; Potu, H.; Kandarpa, M.; Ermann, M.; Courtney, S.M.; Young, M.; Showalter, H.D.; Sun, D.; et al. Targeting deubiquitinase activity with a novel small-molecule inhibitor as therapy for B-cell malignancies. Blood 2015, 125, 3588–3597, doi:10.1182/blood-2014-10-605584.
- Akiyama, H.; Umezawa, Y.; Ishida, S.; Okada, K.; Nogami, A.; Miura, O. Inhibition of USP9X induces apoptosis in FLT3-ITD-positive AML cells cooperatively by inhibiting the mutant kinase through aggresomal translocation and inducing oxidative stress. Cancer Lett. 2019, 453, 84–94, doi:10.1016/j.canlet.2019.03.046.
- Liu, J.; Xia, H.; Kim, M.; Xu, L.; Li, Y.; Zhang, L.; Cai, Y.; Norberg, H.V.; Zhang, T.; Furuya, T.; et al. Beclin1 Controls the Levels of p53 by Regulating the Deubiquitination Activity of USP10 and USP13. Cell 2011, 147, 223–234, doi:10.1016/j.cell.2011.08.037.
- Mistry, H.; Hsieh, G.; Buhrlage, S.J.; Huang, M.; Park, E.; Cuny, G.D.; Galinsky, I.; Stone, R.M.; Gray, N.S.; D’Andrea, A.; et al. Small molecule inhibitors of USP1 target ID1 degradation in leukemic cells. Mol. Cancer Ther. 2013, 12, 2651–2662, doi:10.1158/1535-7163.MCT-13-0103-T.
- Yue, W.; Chen, Z.; Liu, H.; Yan, C.; Chen, M.; Feng, D.; Yan, C.; Wu, H.; Du, L.; Wang, Y.; et al. A small natural molecule promotes mitochondrial fusion through inhibition of the deubiquitinase USP30. Cell Res. 2014, 24, 482–496, doi:10.1038/cr.2014.20.
- Lim, S.-O.; Li, C.-W.; Xia, W.; Cha, J.-H.; Chan, L.-C.; Wu, Y.; Chang, S.-S.; Lin, W.-C.; Hsu, J.-M.; Hsu, Y.-H.; et al. Deubiquitination and Stabilization of PD-L1 by CSN5. Cancer Cell 2016, 30, 925–939, doi:10.1016/j.ccell.2016.10.010.
- Hatcher, H.; Planalp, R.; Cho, J.; Torti, F.M.; Torti, S.V. Curcumin: from ancient medicine to current clinical trials. Cell. Mol. Life Sci. 2008, 65, 1631–1652, doi:10.1007/s00018-008-7452-4.
- Fan, Y.-H.; Cheng, J.; A Vasudevan, S.; Dou, J.; Patel, R.H.; Ma, I.T.; Rojas, Y.; Zhao, Y.; Yu, Y.; Zhang, H.; et al. USP7 inhibitor P22077 inhibits neuroblastoma growth via inducing p53-mediated apoptosis. Cell Death Dis. 2013, 4, e867, doi:10.1038/cddis.2013.400.
- Wang, M.; Zhang, Y.; Wang, T.; Zhang, J.; Zhou, Z.; Sun, Y.; Wang, S.; Shi, Y.; Luan, X.; Zhang, Y.; et al. The USP7 Inhibitor P5091 Induces Cell Death in Ovarian Cancers with Different P53 Status. Cell. Physiol. Biochem. 2017, 43, 1755–1766, doi:10.1159/000484062.
- Hashimoto, M.; Saito, N.; Ohta, H.; Yamamoto, K.; Tashiro, A.; Nakazawa, K.; Inanami, O.; Kitamura, H. Inhibition of ubiquitin‐specific protease 2 causes accumulation of reactive oxygen species, mitochondria dysfunction, and intracellular ATP decrement in C2C12 myoblasts. Physiol. Rep. 2019, 7, e14193, doi:10.14814/phy2.14193.
- Wrigley, J.D.; Eckersley, K.; Hardern, I.M.; Millard, L.; Walters, M.; Peters, S.W.; Mott, R.; Nowak, T.; Ward, R.A.; Simpson, P.B.; et al. Enzymatic Characterisation of USP7 Deubiquitinating activity and Inhibition. Cell Biophys. 2011, 60, 99–111, doi:10.1007/s12013-011-9186-4.
- Sahtoe, D.D.; Sixma, T.K. Layers of DUB regulation. Trends Biochem. Sci. 2015, 40, 456–467, doi:10.1016/j.tibs.2015.05.002.
- Mevissen, T.E.T.; Komander, D. Mechanisms of Deubiquitinase Specificity and Regulation. Annu. Rev. Biochem. 2017, 86, 159–192, doi:10.1146/annurev-biochem-061516-044916.
- Itoh, Y.; Suzuki, M. Design, synthesis, and biological evaluation of novel ubiquitin-activating enzyme inhibitors. Bioorganic Med. Chem. Lett. 2018, 28, 2723–2727, doi:10.1016/j.bmcl.2018.03.004.
- Xu, G.W.; Ali, M.; Wood, T.E.; Wong, D.; Maclean, N.; Wang, X.; Gronda, M.; Skrtic, M.; Li, X.; Hurren, R.; et al. The ubiquitin-activating enzyme E1 as a therapeutic target for the treatment of leukemia and multiple myeloma. Blood 2010, 115, 2251–2259, doi:10.1182/blood-2009-07-231191.
- Tsukamoto, S.; Takeuchi, T.; Rotinsulu, H.; Mangindaan, R.E.; Van Soest, R.W.; Ukai, K.; Kobayashi, H.; Namikoshi, M.; Ohta, T.; Yokosawa, H. Leucettamol A: A new inhibitor of Ubc13-Uev1A interaction isolated from a marine sponge, Leucetta aff. microrhaphis. Bioorganic Med. Chem. Lett. 2008, 18, 6319–6320, doi:10.1016/j.bmcl.2008.10.110.
- Ushiyama, S.; Umaoka, H.; Kato, H.; Suwa, Y.; Morioka, H.; Rotinsulu, H.; Losung, F.; Mangindaan, R.E.P.; De Voogd, N.J.; Yokosawa, H.; et al. Manadosterols A and B, Sulfonated Sterol Dimers Inhibiting the Ubc13–Uev1A Interaction, Isolated from the Marine Sponge Lissodendryx fibrosa. J. Nat. Prod. 2012, 75, 1495–1499, doi:10.1021/np300352u.
- Ceccarelli, D.F.; Tang, X.; Pelletier, B.; Orlicky, S.; Xie, W.; Plantevin, V.; Neculai, D.; Chou, Y.-C.; Ogunjimi, A.; Al-Hakim, A.; et al. An Allosteric Inhibitor of the Human Cdc34 Ubiquitin-Conjugating Enzyme. Cell 2011, 145, 1075–1087, doi:10.1016/j.cell.2011.05.039.
- Sarisozen, C.; Tan, Y.; Liu, J.; Bilir, C.; Shen, L.; Filipczak, N.; Porter, T.M.; Torchilin, V.P.; Filipzcak, N. MDM2 antagonist-loaded targeted micelles in combination with doxorubicin: effective synergism against human glioblastoma via p53 re-activation. J. Drug Target. 2019, 27, 624–633, doi:10.1080/1061186x.2019.1570518.
- Issaeva, N.; Bozko, P.; Enge, M.; Protopopova, M.; Verhoef, L.G.G.C.; Masucci, M.G.; Pramanik, A.; Selivanova, G. Small molecule RITA binds to p53, blocks p53–HDM-2 interaction and activates p53 function in tumors. Nat. Med. 2004, 10, 1321–1328, doi:10.1038/nm1146.
- Shangary, S.; Qin, N.; McEachern, N.; Liu, M.; Miller, R.S.; Qiu, S.; Nikolovska-Coleska, Z.; Ding, K.; Wang, G.; Chen, J.; et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc. Natl. Acad. Sci. USA 2008, 105, 3933–3938, doi:10.1073/pnas.0708917105.
- Li, W.-D.; Wang, M.-J.; Ding, F.; Yin, D.-L.; Liu, Z.-H. Cytotoxic effect of a non-peptidic small molecular inhibitor of the p53-HDM2 interaction on tumor cells. World J. Gastroenterol. 2005, 11, 2927–2931, doi:10.3748/wjg.v11.i19.2927.
- Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Wang, G.; Qiu, S.; Shangary, S.; Gao, W.; Qin, D.; Stuckey, J.; Krajewski, K.; et al. Structure-Based Design of Spiro-oxindoles as Potent, Specific Small-Molecule Inhibitors of the MDM2−p53 Interaction. J. Med. Chem. 2006, 49, 3432–3435, doi:10.1021/jm051122a.
- Bykov, V.J.N.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.M.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288, doi:10.1038/nm0302-282.
- Yang, Y.; Ludwig, R.L.; Jensen, J.P.; Pierre, S.A.; Medaglia, M.V.; Davydov, I.V.; Safiran, Y.J.; Oberoi, P.; Kenten, J.H.; Phillips, A.C.; et al. Small molecule inhibitors of HDM2 ubiquitin ligase activity stabilize and activate p53 in cells. Cancer Cell 2005, 7, 547–559, doi:10.1016/j.ccr.2005.04.029.
- Kitagaki, J.; Agama, K.K.; Pommier, Y.; Yang, Y.; Weissman, A.M. Targeting tumor cells expressing p53 with a water-soluble inhibitor of Hdm2. Mol. Cancer Ther. 2008, 7, 2445–2454, doi:10.1158/1535-7163.MCT-08-0063.
- Herman, A.G.; Hayano, M.; Poyurovsky, M.V.; Shimada, K.; Skouta, R.; Prives, C.; Stockwell, B.R. Discovery of Mdm2-MdmX E3 ligase inhibitors using a cell-based ubiquitination assay. Cancer Discov. 2011, 1, 312–325, doi:10.1158/2159-8290.CD-11-0104.
- Chang, Y.S.; Graves, B.; Guerlavais, V.; Tovar, C.; Packman, K.; To, K.-H.; Olson, K.A.; Kesavan, K.; Gangurde, P.; Mukherjee, A.; et al. Stapled α−helical peptide drug development: A potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc. Natl. Acad. Sci. USA 2013, 110, E3445–E3454, doi:10.1073/pnas.1303002110.
- Wang, H.; Ma, X.; Ren, S.; Buolamwini, J.K.; Yan, C. A small-molecule inhibitor of MDMX activates p53 and induces apoptosis. Mol. Cancer Ther. 2010, 10, 69–79, doi:10.1158/1535-7163.MCT-10-0581.
- Huang, H.-L.; Weng, H.-Y.; Wang, L.-Q.; Yu, C.-H.; Huang, Q.-J.; Zhao, P.-P.; Wen, J.-Z.; Zhou, H.; Qu, L.-H. Triggering Fbw7-Mediated Proteasomal Degradation of c-Myc by Oridonin Induces Cell Growth Inhibition and Apoptosis. Mol. Cancer Ther. 2012, 11, 1155–1165, doi:10.1158/1535-7163.mct-12-0066.
- Orlicky, S.; Tang, X.; Neduva, V.; Elowe, N.; Brown, E.D.; Sicheri, F.; Tyers, M. An allosteric inhibitor of substrate recognition by the SCF(Cdc4) ubiquitin ligase. Nat. Biotechnol. 2010, 28, 733–737, doi:10.1038/nbt.1646.
- Chan, C.-H.; Morrow, J.; Li, C.-F.; Gao, Y.; Jin, G.; Moten, A.; Stagg, L.J.; Ladbury, J.; Cai, Z.; Xu, D.; et al. Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell 2013, 154, 556–568, doi:10.1016/j.cell.2013.06.048.
- Chen, Q.; Xie, W.; Kuhn, D.J.; Voorhees, P.M.; Lopez-Girona, A.; Mendy, D.; Corral, L.G.; Krenitsky, V.P.; Xu, W.; Parseval, L.M.-D.; et al. Targeting the p27 E3 ligase SCFSkp2 results in p27- and Skp2-mediated cell-cycle arrest and activation of autophagy. Blood 2008, 111, 4690–4699, doi:10.1182/blood-2007-09-112904.
- Blees, J.S.; Bokesch, H.R.; Rübsamen, D.; Schulz, K.; Milke, L.; Bajer, M.M.; Gustafson, K.R.; Henrich, C.J.; McMahon, J.B.; Colburn, N.H.; et al. Erioflorin Stabilizes the Tumor Suppressor Pdcd4 by Inhibiting Its Interaction with the E3-ligase β-TrCP1. PLoS ONE 2012, 7, e46567, doi:10.1371/journal.pone.0046567.
- Nakajima, H.; Fujiwara, H.; Furuichi, Y.; Tanaka, K.; Shimbara, N. A novel small-molecule inhibitor of NF-κB signaling. Biochem. Biophys. Res. Commun. 2008, 368, 1007–1013, doi:10.1016/j.bbrc.2008.01.166.
- Aghajan, M.; Jonai, N.; Flick, K.; Fu, F.; Luo, M.; Cai, X.; Ouni, I.; Pierce, N.; Tang, X.; Lomenick, B.; et al. Chemical genetics screen for enhancers of rapamycin identifies a specific inhibitor of an SCF family E3 ubiquitin ligase. Nat. Biotechnol. 2010, 28, 738–742, doi:10.1038/nbt.1645.
- Zeng, X.; Sigoillot, F.; Gaur, S.; Choi, S.; Pfaff, K.L.; Oh, N.-C.; Hathaway, N.; Dimova, N.; Cuny, G.D.; King, R.W. Pharmacologic Inhibition of the Anaphase-Promoting Complex Induces A Spindle Checkpoint-Dependent Mitotic Arrest in the Absence of Spindle Damage. Cancer Cell 2010, 18, 382–395, doi:10.1016/j.ccr.2010.08.010.
- Sackton, K.L.; Dimova, N.; Zeng, X.; Tian, W.; Zhang, M.; Sackton, T.B.; Meaders, J.; Pfaff, K.L.; Sigoillot, F.; Yu, H.; et al. Synergistic blockade of mitotic exit by two chemical inhibitors of the APC/C. Nature 2014, 514, 646–649, doi:10.1038/nature13660.
- Robak, T.; Liu, T.; Samoilova, O.; Pylypenko, H.; Siritanaratkul, N.; Osmanov, E.; Alexeeva, J.; Hong, X.; Okamoto, R.; Pei, L.; et al. Bortezomib-Based Therapy for Newly Diagnosed Mantle-Cell Lymphoma. New Engl. J. Med. 2015, 372, 944–953, doi:10.1056/nejmoa1412096.
- Piva, R.; Ruggeri, B.; Williams, M.; Costa, G.; Tamagno, I.; Ferrero, D.; Giai, V.; Coscia, M.; Peola, S.; Massaia, M.; et al. CEP-18770: A novel, orally active proteasome inhibitor with a tumor-selective pharmacologic profile competitive with bortezomib. Blood 2008, 111, 2765–2775, doi:10.1182/blood-2007-07-100651.
- Zhou, H.-J.; Aujay, M.A.; Bennett, M.K.; Dajee, M.; Demo, S.D.; Fang, Y.; Ho, M.N.; Jiang, J.; Kirk, C.J.; Laidig, G.J.; et al. Design and synthesis of an orally bioavailable and selective peptide epoxyketone Proteasome Inhibitor (PR-047). J. Med. Chem. 2009, 52, 3028–3038, doi:10.1021/jm801329v.
- Chauhan, D.; Singh, A.V.; Aujay, M.; Kirk, C.J.; Bandi, M.; Ciccarelli, B.; Raje, N.; Richardson, P.; Anderson, K.C. A novel orally active proteasome inhibitor ONX 0912 triggers in vitro and in vivo cytotoxicity in multiple myeloma. Blood 2010, 116, 4906–4915, doi:10.1182/blood-2010-04-276626.
- Demo, S.D.; Kirk, C.J.; Aujay, M.A.; Buchholz, T.J.; Dajee, M.; Ho, M.N.; Jiang, J.; Lewis, E.R.; Parlati, F.; Vallone, M.K.; et al. Antitumor Activity of PR-171, a Novel Irreversible Inhibitor of the Proteasome. Cancer Res. 2007, 67, 6383–6391, doi:10.1158/0008-5472.can-06-4086
- Baljevic, M.; Orlowski, R. Pharmacodynamics and pharmacokinetics of proteasome inhibitors for the treatment of multiple myeloma. Expert Opin. Drug Metab. Toxicol. 2019, 15, 459–473, doi:10.1080/17425255.2019.1621839.
- Potts, B.C.; Albitar, M.X.; Anderson, K.C.; Baritaki, S.; Berkers, C.; Bonavida, B.; Chandra, J.; Chauhan, D.; Cusack, J.C.; Fenical, W.; et al. Marizomib, a proteasome inhibitor for all seasons: preclinical profile and a framework for clinical trials. Curr. Cancer Drug Targets 2011, 11, 254–284, doi:10.2174/156800911794519716.
- Fisher, R.I.; Bernstein, S.H.; Kahl, B.S.; Djulbegovic, B.; Robertson, M.J.; De Vos, S.; Epner, E.; Krishnan, A.; Leonard, J.P.; Lonial, S.; et al. Multicenter Phase II Study of Bortezomib in Patients With Relapsed or Refractory Mantle Cell Lymphoma. J. Clin. Oncol. 2006, 24, 4867–4874, doi:10.1200/jco.2006.07.9665.
- Herndon, T.M.; Deisseroth, A.; Kaminskas, E.; Kane, R.C.; Koti, K.M.; Rothmann, M.; Habtemariam, B.; Bullock, J.; Bray, J.D.; Hawes, J.; et al. U.S. Food and Drug Administration Approval: Carfilzomib for the Treatment of Multiple Myeloma. Clin. Cancer Res. 2013, 19, 4559–4563, doi:10.1158/1078-0432.ccr-13-0755.
- Hari, P.; Matous, J.V.; Voorhees, P.M.; Shain, K.H.; Obreja, M.; Frye, J.; Fujii, H.; Jakubowiak, A.J.; Rossi, D.; Sonneveld, P. Oprozomib in patients with newly diagnosed multiple myeloma. Blood Cancer J. 2019, 9, 64–66, doi:10.1038/s41408-019-0232-6.
- Dhakal, B.; D’Souza, A.; Hamadani, M.; Arce-Lara, C.; Schroeder, K.; Chhabra, S.; Shah, N.N.; Gauger, K.; Keaton, T.; Pasquini, M.; et al. Phase I/II trial of bendamustine, ixazomib, and dexamethasone in relapsed/refractory multiple myeloma. Blood Cancer J. 2019, 9, 56, doi:10.1038/s41408-019-0219-3.
- Rowinsky, E.K.; Paner, A.; Berdeja, J.G.; Paba-Prada, C.; Venugopal, P.; Porkka, K.; Gullbo, J.; Linder, S.; Loskog, A.; Richardson, P.G.; et al. Phase 1 study of the protein deubiquitinase inhibitor VLX1570 in patients with relapsed and/or refractory multiple myeloma. Investig. New Drugs 2020, 1–6, doi:10.1007/s10637-020-00915-4.
- U.S. National library of medicine, clinical trials.gov. Available online: https://clinicaltrials.gov/ct2/home (accessed on 15 June 2020)
- Shanmugam, M.K.; Rane, G.; Kanchi, M.M.; Arfuso, F.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; Tan, B.K.H.; Kumar, A.P.; Sethi, G. The Multifaceted Role of Curcumin in Cancer Prevention and Treatment. Molecules 2015, 20, 2728–2769, doi:10.3390/molecules20022728.
- Advani, A.; Cooper, B.W.; Visconte, V.; Elson, P.; Chan, R.; Carew, J.S.; Wei, W.; Mukherjee, S.; Gerds, A.; Carraway, H.E.; et al. A Phase 1/ 2 Trial of MEC (Mitoxantrone, Etoposide, Cytarabine) in Combination with Ixazomib for Relapsed Refractory Acute Myeloid Leukemia. Clin. Cancer Res. 2019, 25, 4231–4237, doi:10.1158/1078-0432.CCR-18-3886.
- Yang, J.; Shi, Y.; Li, C.; Gui, L.; Zhao, X.; Liu, P.; Han, X.; Song, Y.; Li, N.; Du, P.; et al. Phase I clinical trial of pegylated liposomal mitoxantrone plm60-s: pharmacokinetics, toxicity and preliminary efficacy. Cancer Chemother. Pharmacol. 2014, 74, 637–646, doi:10.1007/s00280-014-2523-8.
- Cai, J.; Xia, X.; Liao, Y.; Liu, N.; Guo, Z.; Chen, J.; Yang, L.; Long, H.; Yang, Q.; Zhang, X.; et al. A novel deubiquitinase inhibitor b-AP15 triggers apoptosis in both androgen receptor-dependent and -independent prostate cancers. Oncotarget 2017, 8, 63232–63246, doi:10.18632/oncotarget.18774.
- Kropp, K.N.; Maurer, S.; Rothfelder, K.; Schmied, B.J.; Clar, K.L.; Schmidt, M.; Strunz, B.; Kopp, H.-G.; Steinle, A.; Grünebach, F.; et al. The novel deubiquitinase inhibitor b-AP15 induces direct and NK cell-mediated antitumor effects in human mantle cell lymphoma. Cancer Immunol. Immunother. 2018, 67, 935–947, doi:10.1007/s00262-018-2151-y.
- Jiang, L.; Sun, Y.; Wang, J.; He, Q.; Chen, X.; Lan, X.; Chen, J.; Dou, Q.P.; Shi, X.; Liu, J. Proteasomal cysteine deubiquitinase inhibitor b-AP15 suppresses migration and induces apoptosis in diffuse large B cell lymphoma. J. Exp. Clin. Cancer Res. 2019, 38, 453 doi:10.1186/s13046-019-1446-y.
- Wang, X.; Stafford, W.C.; Mazurkiewicz, M.; Fryknäs, M.; Brjnic, S.; Zhang, X.; Gullbo, J.; Larsson, R.; Arnér, E.S.J.; D’Arcy, P.; et al. The 19S Deubiquitinase Inhibitor b-AP15 Is Enriched in Cells and Elicits Rapid Commitment to Cell Death. Mol. Pharmacol. 2014, 85, 932–945, doi:10.1124/mol.113.091322.
- D’Arcy, P.; Brnjic, S.; Olofsson, M.H.; Fryknäs, M.; Lindsten, K.; De Cesare, M.; Perego, P.; Sadeghi, B.; Hassan, M.; Larsson, R.; et al. Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat. Med. 2011, 17, 1636–1640, doi:10.1038/nm.2536.
- Abramson, H.N. The Multiple Myeloma Drug Pipeline-2018: A Review of Small Molecules and Their Therapeutic Targets. Clin. Lymphoma Myeloma Leuk. 2018, 18, 611–627, doi:10.1016/j.clml.2018.06.015.
- Paulus, A.; Akhtar, S.; Caulfield, T.R.; Samuel, K.; Yousaf, H.; Bashir, Y.; Paulus, S.M.; Tran, D.; Hudec, R.; Cogen, D.; et al. Coinhibition of the deubiquitinating enzymes, USP14 and UCHL5, with VLX1570 is lethal to ibrutinib- or bortezomib-resistant Waldenstrom macroglobulinemia tumor cells. Blood Cancer J. 2016, 6, e492, doi:10.1038/bcj.2016.93.
- Giordano, A.; Tommonaro, G. Curcumin and Cancer. Nutrients 2019, 11, 2376, doi:10.3390/nu11102376.
- Adiwidjaja, J.; McLachlan, A.J.; Boddy, A.V. Curcumin as a clinically-promising anti-cancer agent: pharmacokinetics and drug interactions. Expert Opin. Drug Metab. Toxicol. 2017, 13, 953–972, doi:10.1080/17425255.2017.1360279.
- Bartholomeusz, G.A.; Talpaz, M.; Kapuria, V.; Kong, L.Y.; Wang, S.; Estrov, Z.; Priebe, W.; Wu, J.; Donato, N.J. Activation of a novel Bcr/Abl destruction pathway by WP1130 induces apoptosis of chronic myelogenous leukemia cells. Blood 2007, 109, 3470–3478, doi:10.1182/blood-2006-02-005579.
- Peddaboina, C.; Jupiter, D.; Fletcher, S.; Yap, J.L.; Rai, A.; Tobin, R.P.; Jiang, W.; Rascoe, P.A.; Newell-Rogers, M.K.; Smythe, W.R.; et al. The downregulation of Mcl-1 via USP9X inhibition sensitizes solid tumors to Bcl-xl inhibition. BMC Cancer 2012, 12, 541, doi:10.1186/1471-2407-12-541.
- Wang, S.; Kollipara, R.K.; Srivastava, N.; Li, R.; Ravindranathan, P.; Hernandez, E.; Freeman, E.; Humphries, C.G.; Kapur, P.; Lotan, Y.; et al. Ablation of the oncogenic transcription factor ERG by deubiquitinase inhibition in prostate cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 4251–4256, doi:10.1073/pnas.1322198111.
- Abbi, K.K.S.; Rybka, W.; Ehmann, W.C.; Claxton, D.F. Phase I/II Study of Clofarabine, Etoposide, and Mitoxantrone in Patients With Refractory or Relapsed Acute Leukemia. Clin. Lymphoma Myeloma Leuk. 2015, 15, 41–46, doi:10.1016/j.clml.2014.06.005.
- Gill, H.; Yim, R.; Pang, H.H.; Lee, P.; Chan, T.S.Y.; Hwang, Y.Y.; Leung, G.M.K.; Ip, H.W.; Leung, R.Y.Y.; Yip, S.F.; et al. Clofarabine, cytarabine, and mitoxantrone in refractory/relapsed acute myeloid leukemia: High response rates and effective bridge to allogeneic hematopoietic stem cell transplantation. Cancer Med. 2020, 9, 3371–3382, doi:10.1002/cam4.2865.
- Swords, R.T.; Coutre, S.; Maris, M.B.; Zeidner, J.F.; Foran, J.M.; Cruz, J.; Erba, H.P.; Berdeja, J.G.; Tam, W.; Vardhanabhuti, S.; et al. Pevonedistat, a first-in-class NEDD8-activating enzyme inhibitor, combined with azacitidine in patients with AML. Blood 2018, 131, 1415–1424, doi:10.1182/blood-2017-09-805895.
- Lockhart, A.C.; Bauer, T.M.; Aggarwal, C.; Lee, C.B.; Harvey, R.D.; Cohen, R.B.; Sedarati, F.; Nip, T.K.; Faessel, H.; Dash, A.B.; et al. Phase Ib study of pevonedistat, a NEDD8-activating enzyme inhibitor, in combination with docetaxel, carboplatin and paclitaxel, or gemcitabine, in patients with advanced solid tumors. Invest New Drugs 2019, 37, 87–97, doi:10.1007/s10637-018-0610-0.
- Simon, R.; Freidlin, B.; Rubinstein, L.; Arbuck, S.G.; Collins, J.; Christian, M.C. Accelerated titration designs for phase I clinical trials in oncology. J. Natl. Cancer Inst. 1997, 89, 1138–1147, doi:10.1093/jnci/89.15.1138.
- Navon, A.; Ciechanover, A. The 26 S proteasome: From basic mechanisms to drug targeting. J. Biol. Chem. 2009, 284, 33713–33718.
- Crawford, L.J.; Walker, B.; Irvine, A.E. Proteasome inhibitors in cancer therapy. J. Cell Commun. Signal. 2011, 5, 101–110, doi:10.1007/s12079-011-0121-7.
- Adams, J. The development of proteasome inhibitors as anticancer drugs. Cancer Cell 2004, 5, 417–421, doi:10.1016/s1535-6108(04)00120-5.
- Martin, P.; Ruan, J.; Furman, R.; Rutherford, S.; Allan, J.; Chen, Z.; Huang, X.; DiLiberto, M.; Chen-Kiang, S.; Leonard, J.P. A phase I trial of palbociclib plus bortezomib in previously treated mantle cell lymphoma. Leuk. Lymphoma 2019, 60, 2917–2921, doi:10.1080/10428194.2019.1612062.
- Tomlinson, B.; Tuscano, J.M.; Abedi, M.; Welborn, J.; Arora, M.; O’Donnell, R.T.; Wun, T.; Jonas, B. A phase II study of bortezomib in combination with pegylated liposomal doxorubicin for acute myeloid leukemia. Am. J. Hematol. 2019, 94, E291–E294, doi:10.1002/ajh.25605.
- Huang, I.-T.; Dhungel, B.; Shrestha, R.; Bridle, K.; Crawford, D.H.G.; Jayachandran, A.; Steel, J. Spotlight on Bortezomib: potential in the treatment of hepatocellular carcinoma. Expert Opin. Investig. Drugs 2018, 28, 7–18, doi:10.1080/13543784.2019.1551359.
- O’Connor, O.A.; Stewart, A.K.; Vallone, M.; Molineaux, C.J.; Kunkel, L.A.; Gerecitano, J.F.; Orlowski, R.Z. A phase 1 dose escalation study of the safety and pharmacokinetics of the novel proteasome inhibitor carfilzomib (PR-171) in patients with hematologic malignancies. Clin. Cancer Res. 2009, 15, 7085–7091, doi:10.1158/1078-0432.CCR-09-0822.
- Gu, J.J.; Hernandez-Ilizaliturri, F.J.; Kaufman, G.P.; Czuczman, N.M.; Mavis, C.; Skitzki, J.J.; Czuczman, M.S. The novel proteasome inhibitor carfilzomib induces cell cycle arrest, apoptosis and potentiates the anti-tumour activity of chemotherapy in rituximab-resistant lymphoma. Br. J. Haematol. 2013, 162, 657–669, doi:10.1111/bjh.12452.
- Gupta, S.V.; Hertlein, E.; Lu, Y.; Sass, E.J.; Lapalombella, R.; Chen, T.L.; Davis, M.E.; Woyach, J.A.; Lehman, A.; Jarjoura, D.; et al. The proteasome inhibitor carfilzomib functions independently of p53 to induce cytotoxicity and an atypical NF-κB response in chronic lymphocytic leukemia cells. Clin. Cancer Res. 2013, 19, 2406–2419, doi:10.1158/1078-0432.CCR-12-2754.
- Hasanov, E.; Tidwell, R.S.; Fernandez, P.; Park, L.; McMichael, C.; Tannir, N.M.; Jonasch, E. Phase II Study of Carfilzomib in Patients With Refractory Renal Cell Carcinoma. Clin. Genitourin. Cancer 2019, 17, 451–456, doi:10.1016/j.clgc.2019.07.003.
- Chauhan, D.; Tian, Z.; Zhou, B.; Kuhn, D.; Orlowski, R.; Raje, N.; Richardson, P.; Anderson, K.; Zou, B. In vitro and in vivo selective antitumor activity of a novel orally bioavailable proteasome inhibitor MLN9708 against multiple myeloma cells. Clin. Cancer Res. 2011, 17, 5311–5321, doi:10.1158/1078-0432.CCR-11-0476.
- Rinnerthaler, G.; Gampenrieder, S.P.; Petzer, A.L.; Burgstaller, S.; Fuchs, D.; Rossmann, D.; Balic, M.; Egle, D.; Rumpold, H.; Singer, C.F.; et al. Ixazomib in combination with carboplatin in pretreated women with advanced triple-negative breast cancer, a phase I/II trial of the AGMT (AGMT MBC-10 trial). BMC Cancer 2018, 18, 1074, doi:10.1186/s12885-018-4979-0.
- Sanchorawala, V.; Palladini, G.; Kukreti, V.; Zonder, J.A.; Cohen, A.D.; Seldin, D.C.; Dispenzieri, A.; Jaccard, A.; Schönland, S.O.; Berg, D.; et al. A phase 1/2 study of the oral proteasome inhibitor ixazomib in relapsed or refractory AL amyloidosis. Blood 2017, 130, 597–605, doi:10.1182/blood-2017-03-771220.
- Gupta, N.; Hanley, M.J.; Venkatakrishnan, K.; Wang, B.; Sharma, S.; Bessudo, A.; Hui, A.; Nemunaitis, J. The Effect of a High‐Fat Meal on the Pharmacokinetics of Ixazomib, an Oral Proteasome Inhibitor, in Patients With Advanced Solid Tumors or Lymphoma. J. Clin. Pharmacol. 2016, 56, 1288–1295, doi:10.1002/jcph.719.
- Richardson, P.G.; Zimmerman, T.M.; Hofmeister, C.C.; Talpaz, M.; Chanan-Khan, A.A.; Kaufman, J.L.; Laubach, J.P.; Chauhan, D.; Jakubowiak, A.J.; Reich, S.; et al. Phase 1 study of marizomib in relapsed or relapsed and refractory multiple myeloma: NPI-0052-101 Part 1. Blood 2016, 127, 2693–2700, doi:10.1182/blood-2015-12-686378.
- Levin, N.; Spencer, A.; Harrison, S.J.; Chauhan, D.; Burrows, F.J.; Anderson, K.C.; Reich, S.D.; Richardson, P.G.; Trikha, M. Marizomib irreversibly inhibits proteasome to overcome compensatory hyperactivation in multiple myeloma and solid tumour patients. Br. J. Haematol. 2016, 174, 711–720, doi:10.1111/bjh.14113.
- Harrison, S.J.; Mainwaring, P.; Price, T.; Millward, M.J.; Padrik, P.; Underhill, C.R.; Cannell, P.K.; Reich, S.D.; Trikha, M.; Spencer, A. Phase 1 Clinical Trial of Marizomib (NPI-0052) in Patients with Advanced Malignancies including Multiple Myeloma: Study NPI-0052-102 Final Results. Clin. Cancer Res. 2016, 22, 4559–4566, doi:10.1158/1078-0432.ccr-15-2616.
- Shah, J.; Usmani, S.Z.; Stadtmauer, E.A.; Rifkin, R.M.; Berenson, J.R.; Berdeja, J.G.; Lyons, R.M.; Klippel, Z.; Chang, Y.-L.; Niesvizky, R. Oprozomib, pomalidomide, and Dexamethasone in Patients With Relapsed and/or Refractory Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2019, 19, 570–578, doi:10.1016/j.clml.2019.05.017.
- Ghobrial, I.M.; Vij, R.; Siegel, D.; Badros, A.; Kaufman, J.; Raje, N.; Jakubowiak, A.; Savona, M. R.; Obreja, M.; Berdeja, J.G. A Phase Ib/II Study of Oprozomib in Patients with Advanced Multiple Myeloma and Waldenström Macroglobulinemia. Clin. Cancer Res. 2019, 25, 4907–4916, doi:10.1158/1078-0432.CCR-18-3728.
- Richardson, P.G.; Zimmerman, T.M.; Hofmeister, C.C.; Talpaz, M.; Chanan-Khan, A.A.; Kaufman, J.L.; Laubach, J.P.; Chauhan, D.; Jakubowiak, A.J.; Reich, S.; et al. Phase 1 study of marizomib in relapsed or relapsed and refractory multiple myeloma: NPI-0052-101 Part 1. Blood 2016, 127, 2693–2700, doi:10.1182/blood-2015-12-686378.
- Levin, N.; Spencer, A.; Harrison, S.J.; Chauhan, D.; Burrows, F.J.; Anderson, K.C.; Reich, S.D.; Richardson, P.G.; Trikha, M. Marizomib irreversibly inhibits proteasome to overcome compensatory hyperactivation in multiple myeloma and solid tumour patients. Br. J. Haematol. 2016, 174, 711–720, doi:10.1111/bjh.14113.
- Harrison, S.J.; Mainwaring, P.; Price, T.; Millward, M.J.; Padrik, P.; Underhill, C.R.; Cannell, P.K.; Reich, S.D.; Trikha, M.; Spencer, A. Phase 1 Clinical Trial of Marizomib (NPI-0052) in Patients with Advanced Malignancies including Multiple Myeloma: Study NPI-0052-102 Final Results. Clin. Cancer Res. 2016, 22, 4559–4566, doi:10.1158/1078-0432.ccr-15-2616.
- Shah, J.; Usmani, S.Z.; Stadtmauer, E.A.; Rifkin, R.M.; Berenson, J.R.; Berdeja, J.G.; Lyons, R.M.; Klippel, Z.; Chang, Y.-L.; Niesvizky, R. Oprozomib, pomalidomide, and Dexamethasone in Patients With Relapsed and/or Refractory Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2019, 19, 570–578, doi:10.1016/j.clml.2019.05.017.
- Ghobrial, I.M.; Vij, R.; Siegel, D.; Badros, A.; Kaufman, J.; Raje, N.; Jakubowiak, A.; Savona, M. R.; Obreja, M.; Berdeja, J.G. A Phase Ib/II Study of Oprozomib in Patients with Advanced Multiple Myeloma and Waldenström Macroglobulinemia. Clin. Cancer Res. 2019, 25, 4907–4916, doi:10.1158/1078-0432.CCR-18-3728.


