 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Konstantinos Karakostis | -- | 5283 | 2022-06-09 15:10:27 | | | |
| 2 | Amina Yu | + 78 word(s) | 5361 | 2022-06-10 03:59:29 | | | | |
| 3 | Amina Yu | Meta information modification | 5361 | 2022-06-13 09:31:28 | | |
Video Upload Options
Personalized cancer therapy is a treatment strategy that takes into account the molecular profile of patients in order to stratify them into groups that are more likely to benefit from different therapeutic approaches. Cancer is the second leading cause of death globally. One of the main hallmarks in cancer is the functional deregulation of crucial molecular pathways via driver genetic events that lead to abnormal gene expression, giving cells a selective growth advantage. Driver events are defined as mutations, fusions and copy number alterations that are causally implicated in oncogenesis. Molecular analysis on tissues that have originated from a wide range of anatomical areas has shown that mutations in different members of several pathways are implicated in different cancer types.
1. Personalized Therapeutic Strategies Require Understanding of the Underlying Signaling Pathways’ Mechanisms
Cancer can be regarded as a collection of diseases, all of which share a common feature; the deregulation of key signaling cascades that leads to uncontrolled cellular proliferation [1]. Malignancies arise from alterations in the DNA sequence of genes, as well as from epigenetic changes [2][3][4][5][6]. Both changes induce the activation of oncogenes or the inactivation of tumor-suppressor genes, leading to evasion of growth suppressor function, resistance to cell death (apoptosis), uncontrolled cell cycle, immune evasion as well as increased invasive and metastatic potential [7]. Cancer diagnosis is becoming more reliant on the characterization of mutated pathways that can provide vital information to the separation and clustering of cancer types [8]. Several signaling cascades, including the b-catenin/Wnt, RTK/RAS, p53 and Sonic Hedgehog (SHH) pathways, are linked to cancer formation due to genetic variations that often occur in the respective genes [9][10]. Identification of mutations in genes implicated in such pathways has the potential to reveal the causative driver events of carcinogenesis [11]; it therefore constitutes a topic of paramount importance in precision medicine and for the development of efficient tailor-made drug therapies targeting each cancer type at the molecular level.
2. The p53 Pathway
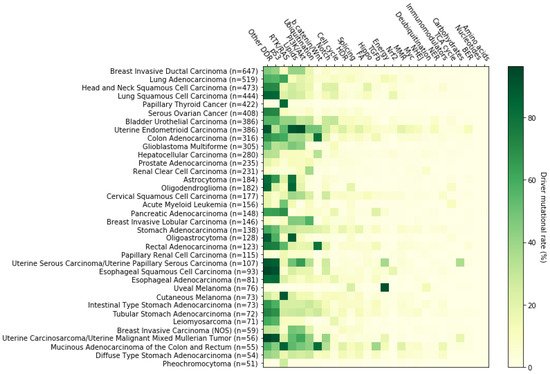
3. The RTK-RAS Pathway
4. Lipid Metabolism
5. The PI3K/AKT Pathway
6. Ubiquitination and Acetylation Pathways
7. The WNT/b Catenin Pathway
8. The Notch Pathway
References
- Parui, A.L.; Bose, K. Cancer Biology and Its Treatment Modalities: A Brief Historical Perspective. In Unravelling Cancer Signaling Pathways: A Multidisciplinary Approach; Bose, K., Chaudhari, P., Eds.; Springer: Singapore, 2019; pp. 1–11.
- Martincorena, I.; Campbell, P.J. Somatic mutation in cancer and normal cells. Science 2016, 349, 1483–1489, Erratum in Science 2016, 351, aaf5401.
- Okugawa, Y.; Grady, W.M.; Goel, A. Epigenetic Alterations in Colorectal Cancer: Emerging Biomarkers. Gastroenterology 2015, 149, 1204–1225.e1212.
- Sever, R.; Brugge, J.S. Signal transduction in cancer. Cold Spring Harb. Perspect. Med. 2015, 5, a006098.
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8.
- Feinberg, A.P.; Koldobskiy, M.A.; Gondor, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e310.
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 789–799.
- Dempke, W.C.M.; Fenchel, K.; Uciechowski, P.; Chevassut, T. Targeting Developmental Pathways: The Achilles Heel of Cancer? Oncology 2017, 93, 213–223.
- Pon, J.R.; Marra, M.A. Driver and passenger mutations in cancer. Annu. Rev. Pathol. 2015, 10, 25–50.
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310.
- Oren, M. Decision making by p53: Life, death and cancer. Cell Death Differ. 2003, 10, 431–442.
- Harris, S.L.; Levine, A.J. The p53 pathway: Positive and negative feedback loops. Oncogene 2005, 24, 2899–2908.
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293.
- Labuschagne, C.F.; Zani, F.; Vousden, K.H. Control of metabolism by p53–Cancer and beyond. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 32–42.
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758.
- Aubrey, B.J.; Strasser, A.; Kelly, G.L. Tumor-Suppressor Functions of the TP53 Pathway. Cold Spring Harb. Perspect. Med. 2016, 6, a026062.
- Haronikova, L.; Olivares-Illana, V.; Wang, L.; Karakostis, K.; Chen, S.; Fahraeus, R. The p53 mRNA: An integral part of the cellular stress response. Nucleic Acids Res. 2019, 47, 3257–3271.
- Muller, P.A.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8.
- Li, J.; Yang, L.; Gaur, S.; Zhang, K.; Wu, X.; Yuan, Y.C.; Li, H.; Hu, S.; Weng, Y.; Yen, Y. Mutants TP53 p.R273H and p.R273C but not p.R273G enhance cancer cell malignancy. Hum. Mutat. 2014, 35, 575–584.
- Sun, S.; Chen, H.; Sun, L.; Wang, M.; Wu, X.; Xiao, Z.J. Hotspot mutant p53-R273H inhibits KLF6 expression to promote cell migration and tumor metastasis. Cell Death Dis. 2020, 11, 595.
- Schulz-Heddergott, R.; Stark, N.; Edmunds, S.J.; Li, J.; Conradi, L.C.; Bohnenberger, H.; Ceteci, F.; Greten, F.R.; Dobbelstein, M.; Moll, U.M. Therapeutic Ablation of Gain-of-Function Mutant p53 in Colorectal Cancer Inhibits Stat3-Mediated Tumor Growth and Invasion. Cancer Cell 2018, 34, 298–314.e297.
- Klemke, L.; Fehlau, C.F.; Winkler, N.; Toboll, F.; Singh, S.K.; Moll, U.M.; Schulz-Heddergott, R. The Gain-of-Function p53 R248W Mutant Promotes Migration by STAT3 Deregulation in Human Pancreatic Cancer Cells. Front. Oncol. 2021, 11, 642603.
- Yu, X.; Vazquez, A.; Levine, A.J.; Carpizo, D.R. Allele-specific p53 mutant reactivation. Cancer Cell 2012, 21, 614–625.
- Grugan, K.D.; Vega, M.E.; Wong, G.S.; Diehl, J.A.; Bass, A.J.; Wong, K.K.; Nakagawa, H.; Rustgi, A.K. A common p53 mutation (R175H) activates c-Met receptor tyrosine kinase to enhance tumor cell invasion. Cancer Biol. Ther. 2013, 14, 853–859.
- Parry, D.; Peters, G. Temperature-sensitive mutants of p16CDKN2 associated with familial melanoma. Mol. Cell Biol. 1996, 16, 3844–3852.
- Rutter, J.L.; Goldstein, A.M.; Davila, M.R.; Tucker, M.A.; Struewing, J.P. CDKN2A point mutations D153spl(c.457G>T) and IVS2+1G>T result in aberrant splice products affecting both p16INK4a and p14ARF. Oncogene 2003, 22, 4444–4448.
- Kannengiesser, C.; Brookes, S.; del Arroyo, A.G.; Pham, D.; Bombled, J.; Barrois, M.; Mauffret, O.; Avril, M.F.; Chompret, A.; Lenoir, G.M.; et al. Functional, structural, and genetic evaluation of 20 CDKN2A germ line mutations identified in melanoma-prone families or patients. Hum. Mutat. 2009, 30, 564–574.
- Angele, S.; Jones, C.; Reis Filho, J.S.; Fulford, L.G.; Treilleux, I.; Lakhani, S.R.; Hall, J. Expression of ATM, p53, and the MRE11-Rad50-NBS1 complex in myoepithelial cells from benign and malignant proliferations of the breast. J. Clin. Pathol. 2004, 57, 1179–1184.
- Bailey, S.L.; Gurley, K.E.; Hoon-Kim, K.; Kelly-Spratt, K.S.; Kemp, C.J. Tumor suppression by p53 in the absence of Atm. Mol. Cancer Res. 2008, 6, 1185–1192.
- Biddlestone-Thorpe, L.; Sajjad, M.; Rosenberg, E.; Beckta, J.M.; Valerie, N.C.; Tokarz, M.; Adams, B.R.; Wagner, A.F.; Khalil, A.; Gilfor, D.; et al. ATM kinase inhibition preferentially sensitizes p53-mutant glioma to ionizing radiation. Clin. Cancer Res. 2013, 19, 3189–3200.
- Malbert-Colas, L.; Ponnuswamy, A.; Olivares-Illana, V.; Tournillon, A.S.; Naski, N.; Fahraeus, R. HDMX folds the nascent p53 mRNA following activation by the ATM kinase. Mol. Cell 2014, 54, 500–511.
- Chwastek, J.; Jantas, D.; Lason, W. The ATM kinase inhibitor KU-55933 provides neuroprotection against hydrogen peroxide-induced cell damage via a gammaH2AX/p-p53/caspase-3-independent mechanism: Inhibition of calpain and cathepsin D. Int. J. Biochem. Cell Biol. 2017, 87, 38–53.
- Karakostis, K.; Vadivel Gnanasundram, S.; Lopez, I.; Thermou, A.; Wang, L.; Nylander, K.; Olivares-Illana, V.; Fahraeus, R. A single synonymous mutation determines the phosphorylation and stability of the nascent protein. J. Mol. Cell Biol. 2019, 11, 187–199.
- Hwang, S.Y.; Kuk, M.U.; Kim, J.W.; Lee, Y.H.; Lee, Y.S.; Choy, H.E.; Park, S.C.; Park, J.T. ATM mediated-p53 signaling pathway forms a novel axis for senescence control. Mitochondrion 2020, 55, 54–63.
- Easton, D.F.; Pharoah, P.D.; Antoniou, A.C.; Tischkowitz, M.; Tavtigian, S.V.; Nathanson, K.L.; Devilee, P.; Meindl, A.; Couch, F.J.; Southey, M.; et al. Gene-panel sequencing and the prediction of breast-cancer risk. N. Engl. J. Med. 2015, 372, 2243–2257.
- Griffith, O.L.; Spies, N.C.; Anurag, M.; Griffith, M.; Luo, J.; Tu, D.; Yeo, B.; Kunisaki, J.; Miller, C.A.; Krysiak, K.; et al. The prognostic effects of somatic mutations in ER-positive breast cancer. Nat. Commun. 2018, 9, 3476.
- Nahar, R.; Zhai, W.; Zhang, T.; Takano, A.; Khng, A.J.; Lee, Y.Y.; Liu, X.; Lim, C.H.; Koh, T.P.T.; Aung, Z.W.; et al. Elucidating the genomic architecture of Asian EGFR-mutant lung adenocarcinoma through multi-region exome sequencing. Nat. Commun. 2018, 9, 216.
- Jette, N.R.; Kumar, M.; Radhamani, S.; Arthur, G.; Goutam, S.; Yip, S.; Kolinsky, M.; Williams, G.J.; Bose, P.; Lees-Miller, S.P. ATM-Deficient Cancers Provide New Opportunities for Precision Oncology. Cancers 2020, 12, 687.
- Zhu, G.; Pan, C.; Bei, J.X.; Li, B.; Liang, C.; Xu, Y.; Fu, X. Mutant p53 in Cancer Progression and Targeted Therapies. Front. Oncol. 2020, 10, 595187.
- Salomao, N.; Karakostis, K.; Hupp, T.; Vollrath, F.; Vojtesek, B.; Fahraeus, R. What do we need to know and understand about p53 to improve its clinical value? J. Pathol. 2021, 254, 443–453.
- Duffy, M.J.; Synnott, N.C.; O’Grady, S.; Crown, J. Targeting p53 for the treatment of cancer. Semin. Cancer Biol. 2020.
- Dhillon, S. Palbociclib: First global approval. Drugs 2015, 75, 543–551.
- Kim, E.S. Abemaciclib: First Global Approval. Drugs 2017, 77, 2063–2070.
- Syed, Y.Y. Ribociclib: First Global Approval. Drugs 2017, 77, 799–807.
- Dhillon, S. Trilaciclib: First Approval. Drugs 2021, 81, 867–874.
- Young, R.J.; Waldeck, K.; Martin, C.; Foo, J.H.; Cameron, D.P.; Kirby, L.; Do, H.; Mitchell, C.; Cullinane, C.; Liu, W.; et al. Loss of CDKN2A expression is a frequent event in primary invasive melanoma and correlates with sensitivity to the CDK4/6 inhibitor PD0332991 in melanoma cell lines. Pigment Cell Melanoma Res. 2014, 27, 590–600.
- Su, D.; Zhang, D.; Jin, J.; Ying, L.; Han, M.; Chen, K.; Li, B.; Wu, J.; Xie, Z.; Zhang, F.; et al. Identification of predictors of drug sensitivity using patient-derived models of esophageal squamous cell carcinoma. Nat. Commun. 2019, 10, 5076.
- DeMichele, A.; Clark, A.S.; Tan, K.S.; Heitjan, D.F.; Gramlich, K.; Gallagher, M.; Lal, P.; Feldman, M.; Zhang, P.; Colameco, C.; et al. CDK 4/6 inhibitor palbociclib (PD0332991) in Rb+ advanced breast cancer: Phase II activity, safety, and predictive biomarker assessment. Clin. Cancer Res. 2015, 21, 995–1001.
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet Oncol. 2015, 16, 25–35.
- Arnedos, M.; Bayar, M.A.; Cheaib, B.; Scott, V.; Bouakka, I.; Valent, A.; Adam, J.; Leroux-Kozal, V.; Marty, V.; Rapinat, A.; et al. Modulation of Rb phosphorylation and antiproliferative response to palbociclib: The preoperative-palbociclib (POP) randomized clinical trial. Ann. Oncol. 2018, 29, 1755–1762.
- Rose, T.L.; Chism, D.D.; Alva, A.S.; Deal, A.M.; Maygarden, S.J.; Whang, Y.E.; Kardos, J.; Drier, A.; Basch, E.; Godley, P.A.; et al. Phase II trial of palbociclib in patients with metastatic urothelial cancer after failure of first-line chemotherapy. Br. J. Cancer 2018, 119, 801–807.
- Toulmonde, M.; Blay, J.Y.; Bouche, O.; Mir, O.; Penel, N.; Isambert, N.; Duffaud, F.; Bompas, E.; Esnaud, T.; Boidot, R.; et al. Activity and Safety of Palbociclib in Patients with Advanced Gastrointestinal Stromal Tumors Refractory to Imatinib and Sunitinib: A Biomarker-driven Phase II Study. Clin. Cancer Res. 2019, 25, 4611–4615.
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708.
- Schmitt, A.; Knittel, G.; Welcker, D.; Yang, T.P.; George, J.; Nowak, M.; Leeser, U.; Buttner, R.; Perner, S.; Peifer, M.; et al. ATM Deficiency Is Associated with Sensitivity to PARP1- and ATR Inhibitors in Lung Adenocarcinoma. Cancer Res. 2017, 77, 3040–3056.
- Lloyd, R.L.; Wijnhoven, P.W.G.; Ramos-Montoya, A.; Wilson, Z.; Illuzzi, G.; Falenta, K.; Jones, G.N.; James, N.; Chabbert, C.D.; Stott, J.; et al. Combined PARP and ATR inhibition potentiates genome instability and cell death in ATM-deficient cancer cells. Oncogene 2020, 39, 4869–4883.
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22.
- Castellano, E.; Molina-Arcas, M.; Krygowska, A.A.; East, P.; Warne, P.; Nicol, A.; Downward, J. RAS signalling through PI3-Kinase controls cell migration via modulation of Reelin expression. Nat. Commun. 2016, 7, 11245.
- Imperial, R.; Toor, O.M.; Hussain, A.; Subramanian, J.; Masood, A. Comprehensive pancancer genomic analysis reveals (RTK)-RAS-RAF-MEK as a key dysregulated pathway in cancer: Its clinical implications. Semin. Cancer Biol. 2019, 54, 14–28.
- Smith, M.J.; Neel, B.G.; Ikura, M. NMR-based functional profiling of RASopathies and oncogenic RAS mutations. Proc. Natl. Acad. Sci. USA 2013, 110, 4574–4579.
- Cook, J.H.; Melloni, G.E.M.; Gulhan, D.C.; Park, P.J.; Haigis, K.M. The origins and genetic interactions of KRAS mutations are allele- and tissue-specific. Nat. Commun. 2021, 12, 1808.
- Feig, L.A.; Cooper, G.M. Relationship among guanine nucleotide exchange, GTP hydrolysis, and transforming potential of mutated ras proteins. Mol. Cell Biol. 1988, 8, 2472–2478.
- Poulin, E.J.; Bera, A.K.; Lu, J.; Lin, Y.J.; Strasser, S.D.; Paulo, J.A.; Huang, T.Q.; Morales, C.; Yan, W.; Cook, J.; et al. Tissue-Specific Oncogenic Activity of KRAS(A146T). Cancer Discov. 2019, 9, 738–755.
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435.
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954.
- Cantwell-Dorris, E.R.; O’Leary, J.J.; Sheils, O.M. BRAFV600E: Implications for carcinogenesis and molecular therapy. Mol. Cancer Ther. 2011, 10, 385–394.
- Falini, B.; Martelli, M.P.; Tiacci, E. BRAF V600E mutation in hairy cell leukemia: From bench to bedside. Blood 2016, 128, 1918–1927.
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075.
- Upadhyaya, M.; Kluwe, L.; Spurlock, G.; Monem, B.; Majounie, E.; Mantripragada, K.; Ruggieri, M.; Chuzhanova, N.; Evans, D.G.; Ferner, R.; et al. Germline and somatic NF1 gene mutation spectrum in NF1-associated malignant peripheral nerve sheath tumors (MPNSTs). Hum. Mutat. 2008, 29, 74–82.
- Brems, H.; Beert, E.; de Ravel, T.; Legius, E. Mechanisms in the pathogenesis of malignant tumours in neurofibromatosis type 1. Lancet Oncol. 2009, 10, 508–515.
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263.
- Zhou, D.; Mattner, J.; Cantu, C., 3rd; Schrantz, N.; Yin, N.; Gao, Y.; Sagiv, Y.; Hudspeth, K.; Wu, Y.P.; Yamashita, T.; et al. Lysosomal glycosphingolipid recognition by NKT cells. Science 2004, 306, 1786–1789.
- Groux-Degroote, S.; van Dijk, S.M.; Wolthoorn, J.; Neumann, S.; Theos, A.C.; De Maziere, A.M.; Klumperman, J.; van Meer, G.; Sprong, H. Glycolipid-dependent sorting of melanosomal from lysosomal membrane proteins by lumenal determinants. Traffic 2008, 9, 951–963.
- Wang, D.; Dubois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193.
- Hishikawa, D.; Hashidate, T.; Shimizu, T.; Shindou, H. Diversity and function of membrane glycerophospholipids generated by the remodeling pathway in mammalian cells. J. Lipid Res. 2014, 55, 799–807.
- Gyamfi, D.; Ofori Awuah, E.; Owusu, S. Chapter 2—Lipid Metabolism: An Overview. In The Molecular Nutrition of Fats; Patel, V.B., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 17–32.
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20.
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033.
- Zhao, L.; Vogt, P.K. Helical domain and kinase domain mutations in p110alpha of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc. Natl. Acad. Sci. USA 2008, 105, 2652–2657.
- Leontiadou, H.; Galdadas, I.; Athanasiou, C.; Cournia, Z. Insights into the mechanism of the PIK3CA E545K activating mutation using MD simulations. Sci. Rep. 2018, 8, 15544.
- Gkeka, P.; Evangelidis, T.; Pavlaki, M.; Lazani, V.; Christoforidis, S.; Agianian, B.; Cournia, Z. Investigating the structure and dynamics of the PIK3CA wild-type and H1047R oncogenic mutant. PLoS Comput. Biol. 2014, 10, e1003895.
- Ikenoue, T.; Kanai, F.; Hikiba, Y.; Obata, T.; Tanaka, Y.; Imamura, J.; Ohta, M.; Jazag, A.; Guleng, B.; Tateishi, K.; et al. Functional analysis of PIK3CA gene mutations in human colorectal cancer. Cancer Res. 2005, 65, 4562–4567.
- Kang, S.; Bader, A.G.; Vogt, P.K. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc. Natl. Acad. Sci. USA 2005, 102, 802–807.
- Bader, A.G.; Kang, S.; Vogt, P.K. Cancer-specific mutations in PIK3CA are oncogenic in vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 1475–1479.
- Dogruluk, T.; Tsang, Y.H.; Espitia, M.; Chen, F.; Chen, T.; Chong, Z.; Appadurai, V.; Dogruluk, A.; Eterovic, A.K.; Bonnen, P.E.; et al. Identification of Variant-Specific Functions of PIK3CA by Rapid Phenotyping of Rare Mutations. Cancer Res. 2015, 75, 5341–5354.
- Rohrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749.
- Snaebjornsson, M.T.; Janaki-Raman, S.; Schulze, A. Greasing the Wheels of the Cancer Machine: The Role of Lipid Metabolism in Cancer. Cell Metab. 2020, 31, 62–76.
- Georgescu, M.M.; Kirsch, K.H.; Akagi, T.; Shishido, T.; Hanafusa, H. The tumor-suppressor activity of PTEN is regulated by its carboxyl-terminal region. Proc. Natl. Acad. Sci. USA 1999, 96, 10182–10187.
- Papa, A.; Wan, L.; Bonora, M.; Salmena, L.; Song, M.S.; Hobbs, R.M.; Lunardi, A.; Webster, K.; Ng, C.; Newton, R.H.; et al. Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell 2014, 157, 595–610.
- Rashmi, R.; DeSelm, C.; Helms, C.; Bowcock, A.; Rogers, B.E.; Rader, J.L.; Rader, J.; Grigsby, P.W.; Schwarz, J.K. AKT inhibitors promote cell death in cervical cancer through disruption of mTOR signaling and glucose uptake. PLoS ONE 2014, 9, e92948.
- Leslie, N.R.; den Hertog, J. Mutant PTEN in Cancer: Worse Than Nothing. Cell 2014, 157, 527–529.
- Salmena, L.; Carracedo, A.; Pandolfi, P.P. Tenets of PTEN tumor suppression. Cell 2008, 133, 403–414.
- Choi, S.W.; Lee, Y.; Shin, K.; Koo, H.; Kim, D.; Sa, J.K.; Cho, H.J.; Shin, H.M.; Lee, S.J.; Kim, H.; et al. Mutation-specific non-canonical pathway of PTEN as a distinct therapeutic target for glioblastoma. Cell Death Dis. 2021, 12, 374.
- Kloosterhof, N.K.; Bralten, L.B.; Dubbink, H.J.; French, P.J.; van den Bent, M.J. Isocitrate dehydrogenase-1 mutations: A fundamentally new understanding of diffuse glioma? Lancet Oncol. 2011, 12, 83–91.
- Clark, O.; Yen, K.; Mellinghoff, I.K. Molecular Pathways: Isocitrate Dehydrogenase Mutations in Cancer. Clin. Cancer Res. 2016, 22, 1837–1842.
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2010, 465, 966.
- Shim, E.H.; Livi, C.B.; Rakheja, D.; Tan, J.; Benson, D.; Parekh, V.; Kho, E.Y.; Ghosh, A.P.; Kirkman, R.; Velu, S.; et al. L-2-Hydroxyglutarate: An epigenetic modifier and putative oncometabolite in renal cancer. Cancer Discov. 2014, 4, 1290–1298.
- Parker, S.J.; Metallo, C.M. Metabolic consequences of oncogenic IDH mutations. Pharmacol. Ther. 2015, 152, 54–62.
- Yang, H.; Ye, D.; Guan, K.L.; Xiong, Y. IDH1 and IDH2 mutations in tumorigenesis: Mechanistic insights and clinical perspectives. Clin. Cancer Res. 2012, 18, 5562–5571.
- Reitman, Z.J.; Jin, G.; Karoly, E.D.; Spasojevic, I.; Yang, J.; Kinzler, K.W.; He, Y.; Bigner, D.D.; Vogelstein, B.; Yan, H. Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc. Natl. Acad. Sci. USA 2011, 108, 3270–3275.
- Esmaeili, M.; Hamans, B.C.; Navis, A.C.; van Horssen, R.; Bathen, T.F.; Gribbestad, I.S.; Leenders, W.P.; Heerschap, A. IDH1 R132H mutation generates a distinct phospholipid metabolite profile in glioma. Cancer Res. 2014, 74, 4898–4907.
- Zhou, L.; Wang, Z.; Hu, C.; Zhang, C.; Kovatcheva-Datchary, P.; Yu, D.; Liu, S.; Ren, F.; Wang, X.; Li, Y.; et al. Integrated Metabolomics and Lipidomics Analyses Reveal Metabolic Reprogramming in Human Glioma with IDH1 Mutation. J. Proteome Res. 2019, 18, 960–969.
- Dumbrava, E.I.; Meric-Bernstam, F. Personalized cancer therapy-leveraging a knowledge base for clinical decision-making. Cold Spring Harb. Mol. Case Stud. 2018, 4, a001578.
- Hudes, G.; Carducci, M.; Tomczak, P.; Dutcher, J.; Figlin, R.; Kapoor, A.; Staroslawska, E.; Sosman, J.; McDermott, D.; Bodrogi, I.; et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 2271–2281.
- Motzer, R.J.; Escudier, B.; Oudard, S.; Hutson, T.E.; Porta, C.; Bracarda, S.; Grunwald, V.; Thompson, J.A.; Figlin, R.A.; Hollaender, N.; et al. Efficacy of everolimus in advanced renal cell carcinoma: A double-blind, randomised, placebo-controlled phase III trial. Lancet 2008, 372, 449–456.
- Benjamin, D.; Colombi, M.; Moroni, C.; Hall, M.N. Rapamycin passes the torch: A new generation of mTOR inhibitors. Nat. Rev. Drug Discov. 2011, 10, 868–880.
- Blair, H.A. Duvelisib: First Global Approval. Drugs 2018, 78, 1847–1853.
- Markham, A. Alpelisib: First Global Approval. Drugs 2019, 79, 1249–1253.
- Dhillon, S. Ivosidenib: First Global Approval. Drugs 2018, 78, 1509–1516.
- Katso, R.; Okkenhaug, K.; Ahmadi, K.; White, S.; Timms, J.; Waterfield, M.D. Cellular function of phosphoinositide 3-kinases: Implications for development, homeostasis, and cancer. Annu. Rev. Cell Dev. Biol. 2001, 17, 615–675.
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619.
- Courtney, K.D.; Corcoran, R.B.; Engelman, J.A. The PI3K pathway as drug target in human cancer. J. Clin. Oncol. 2010, 28, 1075–1083.
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635.
- Cheung, L.W.; Yu, S.; Zhang, D.; Li, J.; Ng, P.K.; Panupinthu, N.; Mitra, S.; Ju, Z.; Yu, Q.; Liang, H.; et al. Naturally occurring neomorphic PIK3R1 mutations activate the MAPK pathway, dictating therapeutic response to MAPK pathway inhibitors. Cancer Cell 2014, 26, 479–494.
- Seton-Rogers, S. Signalling: Uncovering new functions of PI3K mutations. Nat. Rev. Cancer 2014, 14, 766–767.
- Moukarzel, L.A.; Da Cruz Paula, A.; Ferrando, L.; Hoang, T.; Sebastiao, A.P.M.; Pareja, F.; Park, K.J.; Jungbluth, A.A.; Capella, G.; Pineda, M.; et al. Clonal relationship and directionality of progression of synchronous endometrial and ovarian carcinomas in patients with DNA mismatch repair-deficiency associated syndromes. Mod. Pathol. 2021, 34, 994–1007.
- Sun, M.; Hillmann, P.; Hofmann, B.T.; Hart, J.R.; Vogt, P.K. Cancer-derived mutations in the regulatory subunit p85alpha of phosphoinositide 3-kinase function through the catalytic subunit p110alpha. Proc. Natl. Acad. Sci. USA 2010, 107, 15547–15552.
- Li, X.; Lau, A.Y.T.; Ng, A.S.N.; Aldehaiman, A.; Zhou, Y.; Ng, P.K.S.; Arold, S.T.; Cheung, L.W.T. Cancer-associated mutations in the p85alpha N-terminal SH2 domain activate a spectrum of receptor tyrosine kinases. Proc. Natl. Acad. Sci. USA 2021, 118, e2101751118.
- Deng, L.; Wang, C.; Spencer, E.; Yang, L.; Braun, A.; You, J.; Slaughter, C.; Pickart, C.; Chen, Z.J. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 2000, 103, 351–361.
- Pickart, C.M. Ubiquitin in chains. Trends Biochem. Sci. 2000, 25, 544–548.
- Spence, J.; Gali, R.R.; Dittmar, G.; Sherman, F.; Karin, M.; Finley, D. Cell cycle-regulated modification of the ribosome by a variant multiubiquitin chain. Cell 2000, 102, 67–76.
- Huen, M.S.; Grant, R.; Manke, I.; Minn, K.; Yu, X.; Yaffe, M.B.; Chen, J. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 2007, 131, 901–914.
- Mukhopadhyay, D.; Riezman, H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science 2007, 315, 201–205.
- Yumimoto, K.; Akiyoshi, S.; Ueo, H.; Sagara, Y.; Onoyama, I.; Ueo, H.; Ohno, S.; Mori, M.; Mimori, K.; Nakayama, K.I. F-box protein FBXW7 inhibits cancer metastasis in a non-cell-autonomous manner. J. Clin. Investig. 2015, 125, 621–635.
- Yeh, C.H.; Bellon, M.; Nicot, C. FBXW7: A critical tumor suppressor of human cancers. Mol. Cancer 2018, 17, 115.
- Close, V.; Close, W.; Kugler, S.J.; Reichenzeller, M.; Yosifov, D.Y.; Bloehdorn, J.; Pan, L.; Tausch, E.; Westhoff, M.A.; Dohner, H.; et al. FBXW7 mutations reduce binding of NOTCH1, leading to cleaved NOTCH1 accumulation and target gene activation in CLL. Blood 2019, 133, 830–839.
- Koepp, D.M.; Schaefer, L.K.; Ye, X.; Keyomarsi, K.; Chu, C.; Harper, J.W.; Elledge, S.J. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science 2001, 294, 173–177.
- Oberg, C.; Li, J.; Pauley, A.; Wolf, E.; Gurney, M.; Lendahl, U. The Notch intracellular domain is ubiquitinated and negatively regulated by the mammalian Sel-10 homolog. J. Biol. Chem. 2001, 276, 35847–35853.
- Yada, M.; Hatakeyama, S.; Kamura, T.; Nishiyama, M.; Tsunematsu, R.; Imaki, H.; Ishida, N.; Okumura, F.; Nakayama, K.; Nakayama, K.I. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 2004, 23, 2116–2125.
- Wei, W.; Jin, J.; Schlisio, S.; Harper, J.W.; Kaelin, W.G., Jr. The v-Jun point mutation allows c-Jun to escape GSK3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer Cell 2005, 8, 25–33.
- Mao, J.H.; Kim, I.J.; Wu, D.; Climent, J.; Kang, H.C.; DelRosario, R.; Balmain, A. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science 2008, 321, 1499–1502.
- Inuzuka, H.; Shaik, S.; Onoyama, I.; Gao, D.; Tseng, A.; Maser, R.S.; Zhai, B.; Wan, L.; Gutierrez, A.; Lau, A.W.; et al. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature 2011, 471, 104–109.
- Liu, X.; Wang, L.; Zhao, K.; Thompson, P.R.; Hwang, Y.; Marmorstein, R.; Cole, P.A. The structural basis of protein acetylation by the p300/CBP transcriptional coactivator. Nature 2008, 451, 846–850.
- Delvecchio, M.; Gaucher, J.; Aguilar-Gurrieri, C.; Ortega, E.; Panne, D. Structure of the p300 catalytic core and implications for chromatin targeting and HAT regulation. Nat. Struct. Mol. Biol. 2013, 20, 1040–1046.
- Pao, G.M.; Janknecht, R.; Ruffner, H.; Hunter, T.; Verma, I.M. CBP/p300 interact with and function as transcriptional coactivators of BRCA1. Proc. Natl. Acad. Sci. USA 2000, 97, 1020–1025.
- Chan, H.M.; Krstic-Demonacos, M.; Smith, L.; Demonacos, C.; La Thangue, N.B. Acetylation control of the retinoblastoma tumour-suppressor protein. Nat. Cell Biol. 2001, 3, 667–674.
- Grossman, S.R. p300/CBP/p53 interaction and regulation of the p53 response. Eur. J. Biochem. 2001, 268, 2773–2778.
- Friedrichs, N.; Jager, R.; Paggen, E.; Rudlowski, C.; Merkelbach-Bruse, S.; Schorle, H.; Buettner, R. Distinct spatial expression patterns of AP-2alpha and AP-2gamma in non-neoplastic human breast and breast cancer. Mod. Pathol. 2005, 18, 431–438.
- Salloum, R.; McConechy, M.K.; Mikael, L.G.; Fuller, C.; Drissi, R.; DeWire, M.; Nikbakht, H.; De Jay, N.; Yang, X.; Boue, D.; et al. Characterizing temporal genomic heterogeneity in pediatric high-grade gliomas. Acta Neuropathol. Commun. 2017, 5, 78.
- Shi, D.; Pop, M.S.; Kulikov, R.; Love, I.M.; Kung, A.L.; Grossman, S.R. CBP and p300 are cytoplasmic E4 polyubiquitin ligases for p53. Proc. Natl. Acad. Sci. USA 2009, 106, 16275–16280.
- Fahraeus, R.; Olivares-Illana, V. MDM2’s social network. Oncogene 2014, 33, 4365–4376.
- Klein, A.M.; de Queiroz, R.M.; Venkatesh, D.; Prives, C. The roles and regulation of MDM2 and MDMX: It is not just about p53. Genes Dev. 2021, 35, 575–601.
- Attar, N.; Kurdistani, S.K. Exploitation of EP300 and CREBBP Lysine Acetyltransferases by Cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a026534.
- Pasqualucci, L.; Dominguez-Sola, D.; Chiarenza, A.; Fabbri, G.; Grunn, A.; Trifonov, V.; Kasper, L.H.; Lerach, S.; Tang, H.; Ma, J.; et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 2011, 471, 189–195.
- Peifer, M.; Fernandez-Cuesta, L.; Sos, M.L.; George, J.; Seidel, D.; Kasper, L.H.; Plenker, D.; Leenders, F.; Sun, R.; Zander, T.; et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet. 2012, 44, 1104–1110.
- Merk, D.J.; Ohli, J.; Merk, N.D.; Thatikonda, V.; Morrissy, S.; Schoof, M.; Schmid, S.N.; Harrison, L.; Filser, S.; Ahlfeld, J.; et al. Opposing Effects of CREBBP Mutations Govern the Phenotype of Rubinstein-Taybi Syndrome and Adult SHH Medulloblastoma. Dev. Cell 2018, 44, 709–724.e706.
- Mondello, P.; Tadros, S.; Teater, M.; Fontan, L.; Chang, A.Y.; Jain, N.; Yang, H.; Singh, S.; Ying, H.Y.; Chu, C.S.; et al. Selective Inhibition of HDAC3 Targets Synthetic Vulnerabilities and Activates Immune Surveillance in Lymphoma. Cancer Discov. 2020, 10, 440–459.
- Deng, L.; Meng, T.; Chen, L.; Wei, W.; Wang, P. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct. Target. Ther. 2020, 5, 11.
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.K.; Bradner, J.E.; Kaelin, W.G., Jr. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 2014, 343, 305–309.
- Stewart, A.K. Medicine. How thalidomide works against cancer. Science 2014, 343, 256–257.
- Fricker, L.D. Proteasome Inhibitor Drugs. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 457–476.
- Dick, L.R.; Fleming, P.E. Building on bortezomib: Second-generation proteasome inhibitors as anti-cancer therapy. Drug Discov. Today 2010, 15, 243–249.
- Nelson, W.J.; Nusse, R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science 2004, 303, 1483–1487.
- Gattinoni, L.; Zhong, X.S.; Palmer, D.C.; Ji, Y.; Hinrichs, C.S.; Yu, Z.; Wrzesinski, C.; Boni, A.; Cassard, L.; Garvin, L.M.; et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat. Med. 2009, 15, 808–813.
- Petersen, C.P.; Reddien, P.W. Wnt signaling and the polarity of the primary body axis. Cell 2009, 139, 1056–1068.
- Spranger, S.; Gajewski, T.F. A new paradigm for tumor immune escape: Beta-catenin-driven immune exclusion. J. Immunother. Cancer 2015, 3, 43.
- Junge, H.J. Ligand-Selective Wnt Receptor Complexes in CNS Blood Vessels: RECK and GPR124 Plugged In. Neuron 2017, 95, 983–985.
- Miyoshi, Y.; Nagase, H.; Ando, H.; Horii, A.; Ichii, S.; Nakatsuru, S.; Aoki, T.; Miki, Y.; Mori, T.; Nakamura, Y. Somatic mutations of the APC gene in colorectal tumors: Mutation cluster region in the APC gene. Hum. Mol. Genet. 1992, 1, 229–233.
- Fearnhead, N.S.; Britton, M.P.; Bodmer, W.F. The ABC of APC. Hum. Mol. Genet. 2001, 10, 721–733.
- Azzopardi, D.; Dallosso, A.R.; Eliason, K.; Hendrickson, B.C.; Jones, N.; Rawstorne, E.; Colley, J.; Moskvina, V.; Frye, C.; Sampson, J.R.; et al. Multiple rare nonsynonymous variants in the adenomatous polyposis coli gene predispose to colorectal adenomas. Cancer Res. 2008, 68, 358–363.
- Pai, S.G.; Carneiro, B.A.; Mota, J.M.; Costa, R.; Leite, C.A.; Barroso-Sousa, R.; Kaplan, J.B.; Chae, Y.K.; Giles, F.J. Wnt/beta-catenin pathway: Modulating anticancer immune response. J. Hematol. Oncol. 2017, 10, 101.
- Imperial, R.; Ahmed, Z.; Toor, O.M.; Erdogan, C.; Khaliq, A.; Case, P.; Case, J.; Kennedy, K.; Cummings, L.S.; Melton, N.; et al. Comparative proteogenomic analysis of right-sided colon cancer, left-sided colon cancer and rectal cancer reveals distinct mutational profiles. Mol. Cancer 2018, 17, 177.
- Ficari, F.; Cama, A.; Valanzano, R.; Curia, M.C.; Palmirotta, R.; Aceto, G.; Esposito, D.L.; Crognale, S.; Lombardi, A.; Messerini, L.; et al. APC gene mutations and colorectal adenomatosis in familial adenomatous polyposis. Br. J. Cancer 2000, 82, 348–353.
- Mihalatos, M.; Danielides, I.; Belogianni, J.; Harokopos, E.; Papadopoulou, E.; Kalimanis, G.; Tsiava, M.; Triantafillidis, J.K.; Kosmidis, P.A.; Fountzilas, G.; et al. Novel mutations of the APC gene in familial adenomatous polyposis in Greek patients. Cancer Genet. Cytogenet. 2003, 141, 65–70.
- Kitagawa, M.; Hatakeyama, S.; Shirane, M.; Matsumoto, M.; Ishida, N.; Hattori, K.; Nakamichi, I.; Kikuchi, A.; Nakayama, K.; Nakayama, K. An F-box protein, FWD1, mediates ubiquitin-dependent proteolysis of beta-catenin. EMBO J. 1999, 18, 2401–2410.
- Kikuchi, A. Tumor formation by genetic mutations in the components of the Wnt signaling pathway. Cancer Sci. 2003, 94, 225–229.
- Morin, P.J.; Sparks, A.B.; Korinek, V.; Barker, N.; Clevers, H.; Vogelstein, B.; Kinzler, K.W. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 1997, 275, 1787–1790.
- Rubinfeld, B.; Robbins, P.; El-Gamil, M.; Albert, I.; Porfiri, E.; Polakis, P. Stabilization of beta-catenin by genetic defects in melanoma cell lines. Science 1997, 275, 1790–1792.
- Liu, C.; Li, Y.; Semenov, M.; Han, C.; Baeg, G.H.; Tan, Y.; Zhang, Z.; Lin, X.; He, X. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 2002, 108, 837–847.
- Rebouissou, S.; Franconi, A.; Calderaro, J.; Letouze, E.; Imbeaud, S.; Pilati, C.; Nault, J.C.; Couchy, G.; Laurent, A.; Balabaud, C.; et al. Genotype-phenotype correlation of CTNNB1 mutations reveals different ss-catenin activity associated with liver tumor progression. Hepatology 2016, 64, 2047–2061.
- Tu, J.; Park, S.; Yu, W.; Zhang, S.; Wu, L.; Carmon, K.; Liu, Q.J. The most common RNF43 mutant G659Vfs*41 is fully functional in inhibiting Wnt signaling and unlikely to play a role in tumorigenesis. Sci. Rep. 2019, 9, 18557.
- Hao, H.X.; Xie, Y.; Zhang, Y.; Charlat, O.; Oster, E.; Avello, M.; Lei, H.; Mickanin, C.; Liu, D.; Ruffner, H.; et al. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature 2012, 485, 195–200.
- Koo, B.K.; Spit, M.; Jordens, I.; Low, T.Y.; Stange, D.E.; van de Wetering, M.; van Es, J.H.; Mohammed, S.; Heck, A.J.; Maurice, M.M.; et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 2012, 488, 665–669.
- UniProt, C. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489.
- Spit, M.; Fenderico, N.; Jordens, I.; Radaszkiewicz, T.; Lindeboom, R.G.; Bugter, J.M.; Cristobal, A.; Ootes, L.; van Osch, M.; Janssen, E.; et al. RNF43 truncations trap CK1 to drive niche-independent self-renewal in cancer. EMBO J. 2020, 39, e103932.
- Anastas, J.N.; Moon, R.T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 2013, 13, 11–26.
- Jung, Y.S.; Park, J.I. Wnt signaling in cancer: Therapeutic targeting of Wnt signaling beyond beta-catenin and the destruction complex. Exp. Mol. Med. 2020, 52, 183–191.
- Hori, K.; Sen, A.; Artavanis-Tsakonas, S. Notch signaling at a glance. J. Cell Sci. 2013, 126, 2135–2140.
- Bray, S.J. Notch signalling in context. Nat. Rev. Mol. Cell Biol. 2016, 17, 722–735.
- Siebel, C.; Lendahl, U. Notch Signaling in Development, Tissue Homeostasis, and Disease. Physiol. Rev. 2017, 97, 1235–1294.
- Pancewicz, J.; Taylor, J.M.; Datta, A.; Baydoun, H.H.; Waldmann, T.A.; Hermine, O.; Nicot, C. Notch signaling contributes to proliferation and tumor formation of human T-cell leukemia virus type 1-associated adult T-cell leukemia. Proc. Natl. Acad. Sci. USA 2010, 107, 16619–16624.
- Yeh, C.H.; Bellon, M.; Pancewicz-Wojtkiewicz, J.; Nicot, C. Oncogenic mutations in the FBXW7 gene of adult T-cell leukemia patients. Proc. Natl. Acad. Sci. USA 2016, 113, 6731–6736.
- Oswald, F.; Tauber, B.; Dobner, T.; Bourteele, S.; Kostezka, U.; Adler, G.; Liptay, S.; Schmid, R.M. p300 acts as a transcriptional coactivator for mammalian Notch-1. Mol. Cell Biol. 2001, 21, 7761–7774.
- Wallberg, A.E.; Pedersen, K.; Lendahl, U.; Roeder, R.G. p300 and PCAF act cooperatively to mediate transcriptional activation from chromatin templates by notch intracellular domains in vitro. Mol. Cell Biol. 2002, 22, 7812–7819.
- Huang, Y.H.; Cai, K.; Xu, P.P.; Wang, L.; Huang, C.X.; Fang, Y.; Cheng, S.; Sun, X.J.; Liu, F.; Huang, J.Y.; et al. CREBBP/EP300 mutations promoted tumor progression in diffuse large B-cell lymphoma through altering tumor-associated macrophage polarization via FBXW7-NOTCH-CCL2/CSF1 axis. Signal Transduct. Target. Ther. 2021, 6, 10.
- Majumder, S.; Crabtree, J.S.; Golde, T.E.; Minter, L.M.; Osborne, B.A.; Miele, L. Targeting Notch in oncology: The path forward. Nat. Rev. Drug Discov. 2021, 20, 125–144.
- Pannuti, A.; Foreman, K.; Rizzo, P.; Osipo, C.; Golde, T.; Osborne, B.; Miele, L. Targeting Notch to target cancer stem cells. Clin. Cancer Res. 2010, 16, 3141–3152.
- Takebe, N.; Nguyen, D.; Yang, S.X. Targeting notch signaling pathway in cancer: Clinical development advances and challenges. Pharmacol. Ther. 2014, 141, 140–149.

