1. Personalized Therapeutic Strategies Require Understanding of the Underlying Signaling Pathways’ Mechanisms
Cancer can be regarded as a collection of diseases, all of which share a common feature; the deregulation of key signaling cascades that leads to uncontrolled cellular proliferation [7]. Malignancies arise from alterations in the DNA sequence of genes, as well as from epigenetic changes [8,9,10,11,12]. Both changes induce the activation of oncogenes or the inactivation of tumor-suppressor genes, leading to evasion of growth suppressor function, resistance to cell death (apoptosis), uncontrolled cell cycle, immune evasion as well as increased invasive and metastatic potential [13]. Cancer diagnosis is becoming more reliant on the characterization of mutated pathways that can provide vital information to the separation and clustering of cancer types [14]. Several signaling cascades, including the b-catenin/Wnt, RTK/RAS, p53 and Sonic Hedgehog (SHH) pathways, are linked to cancer formation due to genetic variations that often occur in the respective genes [15,16]. Identification of mutations in genes implicated in such pathways has the potential to reveal the causative driver events of carcinogenesis [17]; it therefore constitutes a topic of paramount importance in precision medicine and for the development of efficient tailor-made drug therapies targeting each cancer type at the molecular level.
2. The p53 Pathway
Components of the p53 pathway function cooperatively to ensure that the cell will respond effectively against a variety of stress signals that threaten cellular homeostasis and genomic integrity. In this process, p53 target genes trigger several pathways linked to cancer, such as cell cycle arrest, senescence or apoptosis, blood vessel formation and the regulation of metabolism [
30,
31,
32,
33,
34]. The importance of
TP53 in cancer development is evident by the fact that it occupies the first place in the ranking of genes found to be most frequently altered in cancer [
35,
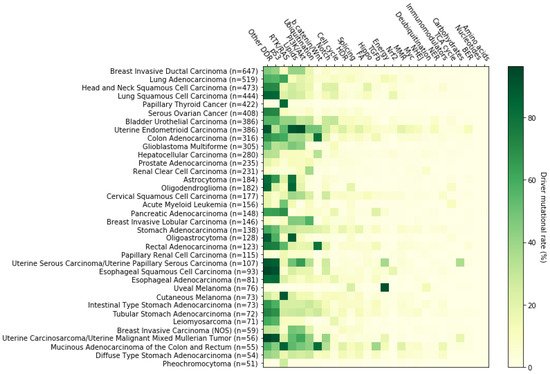
36]. Indeed, the number of cancer types/subtypes where p53 pathway is likely not perturbed is very small (e.g., uveal melanoma) (
Figure 1). In the majority of cancer types, more than 30% of the samples examined harbored driver mutations in at least one key gene of this pathway, while in some cases this percentage exceeded 90% (e.g., esophageal squamous cell carcinoma). Even though there is a high number of cancers that bear a variant in one or more components of the p53 pathway, the affected components are specific to three genes in this pathway. We found that among those patients, 93.66% had a driver mutation in
TP53, whereas
CDKN2A and
ATM genes were affected in 7.71% and 6.27% respectively, dictating the simultaneous presence of some of these mutations in a number of cases.
TP53 driver mutations can be located in virtually any region of the gene, thus affecting the ability of the p53 protein to appropriately interact with either its protein effectors or the DNA [
37]. According to our analysis, arginine residues on positions 273, 248 and 175, all of which belong to the DNA-binding domain [
38], are the most frequently mutated, representing 7.06%, 5.87% and 4.41% of patients that have an affected
TP53 gene, respectively. R273 variants have been associated with an increased proliferative and invasive potential, which in the case of R273H, are induced by the inhibition of tumor suppressor
KLF6 [
39,
40]. Similar effects seem to take place when a R248 mutant is present, but this particular mutation leads to a robust binding of certain variants—mainly the mutp53
R248Q—to the STAT3 protein, and to the subsequent activation of the latter [
41,
42]. The R175H variant, on the other hand, is associated with a radical change in p53 conformation which triggers the transactivation of a panel of genes and results in elevated c-met protein levels and to the induction of tumor invasion, among other events [
43,
44].
Figure 1. Alteration rate of 27 signaling pathways across 36 cancer types/subtypes. For this analysis, 10,066 tumor samples of primary origin and with available mutational profiles were examined for the presence of somatic driver mutations in our 204 genes of interest. Calculations were performed by taking into account all the available mutationally profiled primary tumor samples of the cBioPortal selected studies (10,066 samples) instead of the driver event-harboring mutationally profiled primary tumor samples (7915 samples). If for a selected cancer type there were x mutationally profiled primary tumor samples available in the cBioPortal selected studies, then, y samples would bear at least one driver event in at least one of the 204 genes of interest and z samples would harbor driver events in genes participating in a selected signaling pathway. The corresponding driver mutational rate for this cancer type-signaling pathway pair is calculated as (z/x) × 100. Here, only cancer types/subtypes that entail more than 50 analyzed samples are shown. From left to right, signaling pathways are displayed in descending order of total mutational frequency. n: number of samples examined.
In addition to missense mutations and other mutation types, the
CDKN2A gene is frequently affected by nonsense mutations, in line with our analysis, which demonstrates that R80*, R58*, W110*, E120*, Y44* and E88* together constitute almost a third of cancer cases that harbor a defective
CDKN2A gene. Such alterations shut down one of the cell cycle’s checkpoints, as they force the p16
INK4a protein to lose its Cdk-binding ability, thereby being unable to prevent the G1/S transition [
45,
46,
47]. From the remaining two thirds of cases, an aberrant splicing involving the D153 residue appears in 5.16% of all
CDKN2A affected cancer patients. Changes in this position may inactivate both
CDKN2A gene products, i.e., p16
INK4a and p14
ARF, leading to loss of cell cycle control via both pRB and p53 inactivation [
46].
Factors upstream of p53 are also affected and, along with the ATM kinase, play a vital role in the activation of p53 in response to DNA damage [
48,
49,
50,
51,
52,
53,
54]. In our dataset, non-sense mutations leading to premature truncated forms of the ATM protein product represented more than one third of all
ATM mutated cases, with R250* being the main representative. In these cases, the final gene product loses its functionality as a main DNA damage sensor, either partially or completely, thus increasing the vulnerability of the cell to malignant transformation [
55]. Our analysis has also revealed that R337C/H are the most common cancer-related missense mutations in the
ATM gene, accounting together for 7.94% of all
ATM variant carriers. Although these mutations have been documented in cancer patients [
56,
57], to the best of our knowledge their functional impact has not been elucidated yet [
58].
As expected, the dynamic presence of these genes on the mutational cancer map, has made the p53 pathway—and especially the
TP53 component—an attractive therapeutic target. Among a plethora of p53-targeting strategies, the most promising fall into one of the following categories: (i) restoring the function of p53 protein or (ii) impeding the interaction between p53 and its main negative regulator, the MDM2 E3 ubiquitin ligase [
35,
59,
60]. In this context, eprenetapopt (APR-246), a mutant-p53 conformation resetting agent, is probably the most promising compound [
61] and the only therapy of this category that is currently being tested in a phase III clinical trial (NCT03745716); on the other hand, a phase III trial of idasanutlin (NCT02545283), another emerging MDM2 inhibitor, was recently terminated due to low efficacy. The results of such clinical trials, as well as the efforts to improve the properties of the relevant compounds are highly anticipated. Apart from p53, there are four FDA approved anticancer drugs targeting
CDKN2A signaling: abemaciclib, palbociclib, ribociclib and trilaciclib [
62,
63,
64,
65]. All these therapeutic agents act as CDK4/6 inhibitors, aiming to restore the inactivated
CDKN2A [
66,
67]. However, it has been noted that p16
ΙΝΚ4a loss does not necessarily predict response to CDK4/6 inhibitors [
68,
69,
70,
71,
72]. The rationale behind
ATM deficient-related therapeutic approaches however, is to cause synthetic lethality. There is evidence that this is feasible by further weakening DNA repair mechanisms via administration of appropriate inhibitors, mainly PARP and ATR inhibitors, either alone or in combination [
73,
74,
75]. Ongoing clinical trials will validate the potential of preliminary preclinical trial results and their translation into clinical practice.
3. The RTK-RAS Pathway
RTK-RAS is probably the most thoroughly studied cancer-related signaling pathway. Its involvement in a multitude of crucial physiological processes, such as cell growth, proliferation, differentiation, angiogenesis, integrin signaling and cell migration [
76,
77,
78], among many others, makes it clear that deregulation of this pathway can facilitate both tumor initiation and tumor progression. Indeed, our analysis showed that in 12 of 36 cancer types and subtypes examined, more than 30% of patients carried driver mutations in RTK-RAS pathway genes, with this percentage rising above 80% in papillary thyroid cancer, cutaneous melanoma and mucinous adenocarcinoma of the colon and rectum. On the other hand, as with the p53 pathway, no uveal melanoma patient was found to be affected (
Figure 1). The predominantly mutated genes involved in this pathway, i.e.,
KRAS,
BRAF and
NF1, were found mutated in 26.05%, 17.06% and 13.06% of RTK-RAS perturbed cases respectively, while additional mutations in all cancer-related receptor tyrosine kinase-encoding genes represented 33.2% of all mutations. In descending order of mutational frequency, these were as follows:
EGFR,
ERBB2,
FGFR3,
FLT3,
FGFR2,
ERBB3,
RET,
KIT,
MET,
ERBB4,
PDGFRA,
ALK,
FGFR1,
NTRK3,
NTRK1,
ROS1,
NTRK2,
IGF1R and
FGFR4.
Ιn the cohort, we ascertained that
KRAS is almost exclusively mutated (missense mutations) on the G12, G13, Q61 and A146 residues, corresponding to more than 95% of all cases (75.41%, 11.68%, 5.84% and 3.67%, respectively). Mutants of codon 12, 13 and 61 decrease the GTP-hydrolysis rate, with mutants of codon 12 and 16 also being capable of increasing the rate of GDP-exchange [
79,
80], while mutants of codon 146 act mainly through the second mechanism [
81,
82]. Both mechanisms lead to increased levels of the activated GTP-bound form of the KRAS protein, resulting in uncontrolled mitogenic processes via a constitutively active signal transduction [
80,
83].
V600 is the most prevalent mutated codon of the
BRAF gene, occurring in 81.74% of all
BRAF mutated primary tumors in our dataset, while rearrangement of
BRAF with a variety of genes seemed to occur in only 4.36% of these samples. In the majority of cases where a V600 variant exists, the valine residue is substituted by glutamic acid (V600E), enhancing the serine/threonine kinase activity of BRAF, thus leading to the abrogation of extracellular signals, promoting cell growth and proliferation [
84,
85,
86].
In contrast to
BRAF, there is no hotspot position for the
NF1 gene. The most common somatic mutation of
NF1, namely R2450*, barely exceeds 3% of all identified variants in our cohort. In addition, our analysis showed that nonsense mutations, frameshift deletions and splice-site variants are the most frequent mutation types for this gene in cancer, accounting for 39.91%, 20.83% and 15.79% of all identified variants, respectively. Regardless of the mutation type, in the majority of
NF1-affected cases, mutations lead to a loss of neurofibromin GTPase-activating function, resulting in sustainably high GTP-bound levels of RAS proteins and thereby in a continuous tumor-promoting signaling process via RTK-RAS and PI3K/AKT pathway hyperactivation [
87,
88,
89,
90].
4. Lipid Metabolism
Both lipid synthesis and catabolism are essential for the maintenance of membrane functionality, protein trafficking, immune cell responses, signaling, as well as coverage of metabolic demands, such as energy production and storage [
101,
102,
103,
104,
105]. It has been shown that cancer cells manage to retain a high proliferative potential, through various metabolic modifications [
13,
106,
107]. According to our analysis, more than 40% of patients of one in three cancer types/subtypes are characterized by driver mutations in genes involved in lipid metabolism. In uterine endometrioid carcinoma, astrocytoma, oligodendroglioma and oligoastrocytoma this proportion reached or exceeded 80%, while, as with the two pathways discussed above, no primary uveal melanoma tumor of our dataset was affected (
Figure 3). At the gene level,
PIK3CA,
PTEN and
IDH1 occupy the first places in the mutagenicity ranking, as they were found altered in 45.21%, 27.56% and 16.93% of the patients, respectively. Over 95% of
PIK3CA somatic alterations in cancer are missense mutations, half of which include three substitutions: E545K, H1047R and E542K. These substitutions exert a gain-of-function effect on the PI3K protein via two different mechanisms. Amino acid (aa) residues E545 and E542 are located in the helical domain of p110alpha protein and their substitution by a lysine residue attenuates the inhibitory interaction between the catalytic subunit (p110alpha) and the nSH2 domain of the regulatory subunit (p85alpha) of the PI3K protein, promoting the continuous activation of PI3K [
108,
109]. A constitutive PI3K activation is also the result of the kinase domain variant H1047R, but in this case, p110alpha abolishes its C-terminal tail self-inhibitory capacity [
110]. Both mechanisms lead to a strong activation of PI3K downstream effectors, such as the AKT and P70S6K proteins, which mediate protein synthesis, cell growth, cell proliferation, angiogenesis, survival, and thus contribute to tumorigenic transformation [
111,
112,
113,
114]. At the same time, hyper-activated PI3K/AKT signaling induces the expression of genes involved in fatty acid anabolism, a process that generates the essential building blocks for the synthesis of new cells [
115,
116].
PTEN alterations show greater variety than
PIK3CA alterations, with missense mutations, frameshift insertion-deletions (indels) and nonsense mutations representing 91% of all carriers. The most prevalent mutations involve R130, R233 and T319 aa residues and are found in 17.5%, 5% and 4.74% of all affected patients, respectively. In the majority of cases, arginine in position 130 is either substituted by a glutamine or a glycine residue, or, as in the cases of R233 and T319, the codon that is responsible for encoding it, is converted to a stop codon. Whereas truncating mutations such as R130*, R233* and T319* lead to an unstable, non-functional protein product [
117,
118,
119], R130Q and R130G variants generate stable proteins, which, however, lack phosphatase activity [
118,
120]. Absence or non-functionality of the PTEN protein prevents PIP3 dephosphorylation, which in turn accumulates and recruits PI3K signaling effectors, such as AKT and PDK1 [
121]. Furthermore, it was recently shown that PTEN
R130Q mutants tend to accumulate at the cell periphery where they form leading edges that increase tumor invasiveness and further activate the PI3K/AKT signaling axis [
122].
A very impressive paradigm of a predominant mutational hotspot is represented by the
IDH1 gene. Of the 467 somatic driver mutations that we identified in an equal number of cancer patients, 466 carried an R132 replacement, with histidine being the most frequent substitute (388/466 cases); cysteine, glycine, serine and leucine substitutions occurred in 50, 16, 11 and one patients, respectively. These variants, which are mapped in the catalytic pocket of the enzyme, make isocitrate dehydrogenase-1 convert the normal final product of its catalytic activity, alpha-ketoglutarate (aKG), into R(−)-2-hydroxyglutarate (2HG), with concomitant NADPH consumption [
123,
124]. The following 2HG-mediated inhibition of aKG-dependent deoxygenases, such as TET2 and JMJD2A/C, promotes global gene expression changes, which along with the redox stress arising from the declined NADPH levels, reflect the tumorigenic impact of these mutations [
124,
125,
126,
127]. Likewise, even though the decreased levels of two lipogenesis components, NADPH and aKG [
128] would be expected to abrogate lipid synthesis, certain lipid precursors, such as glycerol-phosphates and glycerophosphocholine, are present in elevated quantities; on the other hand, other lipid precursors, as for example myo-inositol phosphate, are present in reduced levels, compared to unaffected tumors, suggesting that cancer cells harboring
IDH1 variants, alter their phospholipid expression profile, probably in a tumor-assisting manner [
129,
130,
131].
As made clear from the above, signaling changes involving lipid metabolism regulators perturb a wide spectrum of cellular processes and contribute to cancer development. This observation has led to the development of therapies that mitigate these changes. Currently, there are eleven clinically available therapies that target the signaling of four out of the 24 lipid metabolism-related genes found mutated in our dataset [
91]. As our analysis suggests, when lipid metabolism is deregulated on account of a genetic change, this alteration invariably affects one of its aforementioned PI3K/AKT signaling-mediating regulators. Hence, inhibition of the PI3K/AKT signaling axis is suggested to be the main goal of personalized medicine for the treatment of tumors bearing such alterations. When PI3K catalytic subunit inhibitors, both isoform-specific, such as alpelisib, and pan-isoform, such as copanlisib and duvelisib, as well as selective MTOR inhibitors, including everolimus and temsirolimus, are employed, they seem to offer a significant PFS—or even OS—benefit to patients with a variety of blood or solid malignancies [
132,
133,
134,
135,
136]. In addition, ivosidenib, an inhibitor of
IDH1R132 mutants, has been approved by the FDA, for the treatment of acute myeloid leukemia (AML) patients harboring these variants [
137], while its effectiveness in other
IDH1-deficient cancer types is currently being tested in several ongoing clinical trials (e.g., advanced and metastatic cholangiocarcinoma/NCT02989857, chondrosarcoma/NCT04278781, cholangiocarcinoma—chondrosarcoma—glioma—other advanced solid tumors/NCT02073994, recurrent ependymoma—recurrent ewing sarcoma—recurrent hepatoblastoma/NCT04195555, etc.).
5. The PI3K/AKT Pathway
The impact of the PI3K/AKT signaling network on diverse cellular functions is well established. Many of these, including cell growth and proliferation, survival, motility, cellular metabolism, immune system functions and angiogenesis, are tightly intertwined with cancer development and progression and as a result a lot of research has been dedicated to unraveling the deregulation mechanisms of this pathway in cancer [
138,
139,
140,
141]. In 26 of the 36 cancer types of our dataset, more than 10% of patients carried at least one driver mutation in genes involved in this pathway. Uterine malignancies showed the highest mutational rates with 93.3% of uterine endometrioid carcinoma patients being affected, while the corresponding proportion of the remaining uterine tumors analyzed exceeded 60%. On the other hand, less than 5% of serous ovarian cancer, papillary thyroid cancer, pheochromocytoma, uveal melanoma and acute myeloid leukemia patients appeared to bear such changes (
Figure 1). Among all PI3K/AKT-deregulated tumors examined, 53.75% harbored driver mutations in
PIK3CA, 32.76% in
PTEN and 12.93% in
PIK3R1, with the rest of the genes in this pathway being altered in less than 6% of the tumors each. Interestingly, simultaneous mutations in at least two, or in certain cases in all of the above mentioned top mutated genes, were present in 16.34% of these patients. As driver mutations and therapeutic applications regarding
PIK3CA and
PTEN genes were discussed earlier in this report, we will now focus on the
PIK3R1 gene. Somatic mutations in
PIK3R1 exhibit a highly scattered pattern. The absence of predominant hotspots is reflected by the mutational rates of the most prevalent genetic changes of this gene; 6.67% for R348*, 5.67% for X582_splice and 4% for G376R. R348* is a truncating but gain-of-function mutation that exerts its tumorigenic impact in both a PI3K/AKT-dependent and a PI3K/AKT-independent manner. In addition to activating PI3K/AKT signaling, p85alpha mutants are localized into the nucleus and promote the activation of ERK and JNK kinases, thereby inhibiting FASL-mediated apoptosis and inducing cell survival, growth, proliferation and invasion [
142,
143]. Even though X582 splice alteration has been previously identified and considered as pathogenic [
144], its exact functional consequences have not been elucidated yet. Finally, the nSH2 domain-located G376R substitution acts in the same way as the previously discussed E545 and E542 substitutions of the p110alpha protein, thus attenuating the inhibitory interaction between the regulatory and the catalytic subunit of the PI3K complex and enhancing the PI3K/AKT signaling [
145,
146].
Overall, the PI3K/AKT signaling-promoting behavior of p85alpha mutants, places PIK3R1-targeting in the same therapeutic context as the other major effectors of this pathway, PIK3CA and PTEN.
6. Ubiquitination and Acetylation Pathways
Ubiquitination is a reversible modification that leads either to protein degradation or to the regulation of protein–protein interactions. It is essential for the appropriate execution of various cellular events, namely inflammation, translation, endocytosis, DNA damage response (DDR), protein trafficking, differentiation and signal transduction [
150,
151,
152,
153,
154]. Thus, deregulation of the ubiquitin pathway can lead to cancer initiation and/or progression. Mutations in genes encoding components or regulators of the ubiquitination machinery are not uncommon in cancer samples. Our analysis demonstrates that such mutations are present in more than 10% of patients, in two out of three cancer types (24/36). The highest rates are shown in uterine carcinosarcoma, mucinous adenocarcinoma of the colon and rectum, uterine endometrioid carcinoma and breast invasive lobular carcinoma, ranging from ~42% to ~57%. Contrariwise, oligodendroglioma, papillary thyroid cancer, leiomyosarcoma, uveal melanoma and pheochromocytoma, exhibited the lowest rates with less than 3% of the patients being affected (
Figure 3). At the gene level, the most frequently mutated genes,
FBXW7 (or
FBW7),
EP300 and
CREBBP (or
CBP) were detected in 19.6%, 10.72% and 9.88%, respectively, of ubiquitination/deubiquitination-deficient tumors, while the mutational rate of nine more genes ranged between ~5% and ~9%. In descending order of mutational frequency, these are as follows:
KMT2B,
RNF43,
VHL,
MAP3K1,
KMT2A,
SPOP,
BAP1,
KEAP1 and
BRCA1.
Three arginine residues of the FBW7 ubiquitin ligase—R465, R505 and R479—represent 43.59% of all FBW7 somatic driver mutations in primary tumor samples. These positions are located into the WD40 domain, which constitutes the substrate-binding site of the SCF complex (a type of E3 ligase), which is responsible for the ubiquitin-labeling and the subsequent proteasome-mediated degradation of its protein effectors [
155,
156]. These mutations induce changes in the WD40 domain, altering the interaction potential of FBW7, probably via both hydrophobic and electrostatic interactions, as well as by limiting the contact surface because of the shorter substitute sidechains (mainly cysteine, histidine, glycine and glutamine) [
157]. Given that several FBW7 interactors act as regulators of cell growth, apoptosis and proliferation [
158,
159,
160,
161,
162,
163]; prevention of their degradation may result in tumorigenesis.
Even though the
EP300 gene is usually altered by nonsense mutations, the most recurrent mutations fall into three other categories. The D1399N substitution is the most prevalent somatic mutation of this gene, accounting for 7.29% of
EP300-mutated primary tumors in our working dataset. Missense mutations in this position were shown to change the conformation of the p300 protein histone acetyltransferase (HAT) domain, leading to abolishment of its autoacetylation activity, which is essential for appropriate function [
164,
165]. The subsequent inability of p300 to stimulate other tumor suppressors in the nucleus, such as RB1, BRCA1, p53 and AP-2alpha [
166,
167,
168,
169,
170], paves the way for the predominance of its spatial distinct, cytoplasmic ubiquitin ligase activity, which targets p53 for degradation [
171,
172,
173]. These changes, together with the reduced global levels of histone H3 acetylation [
174], contribute to the tumorigenic impact of this genetic alteration. The second and third most prevalent mutations of the
EP300 gene are the frameshift deletion M1470Cfs*26 and the splice site variant X1429 splice, and were each identified in 2.6% of
EP300-affected patients. However, their functional impact is not clear. Another gene with intrinsic histone acetyltransferase activity, sharing high similarity to
EP300, is the
CREBBP gene. Missense mutations involving the R1446 are the most recurrent somatic mutations of
CREBBP, followed by frameshift indels involving I1084 and substitutions of D1435, accounting for 7.91%, 5.65% and 2.82% of
CREBBP-affected primary tumors, respectively. Both R1446 and D1435 are located in the HAT domain, which is responsible for the catalytic activity of the CBP transcriptional coactivator. Substitution of these aa residues, reduces the acetyl-CoA binding affinity of CBP, thereby impairing its acetyltransferase activity [
175,
176,
177]. Consequently, CBP can neither activate p53 tumor suppressor nor inactivate BCL6 proto-oncoprotein. Furthermore, as with its structurally and functionally related p300 protein, CBP also exhibits an E4 ubiquitin ligase activity targeting p53 for degradation in the cytoplasm [
171]. These observations, in conjunction with the arisen extended transcriptional changes, dictate a tumor promoting effect for these mutations [
175,
178]. The functional impact of frameshift deletion I1084Sfs*15, which is found in 4.52% of
CREBBP-altered samples thus far remains obscure.
Proteins involved in either the attachment or the removal of ubiquitin moieties are barely exploited in clinical practice nowadays [
179]. An exception to this rule is provided by thalidomide analogues lenalidomide and pomalidomide, which have been approved for the treatment of various blood malignancies and exhibit anti-tumor activities by altering the specificity of cereblon, which constitutes the substrate recognition component of the CRL4 E3 ubiquitin ligase complex [
180,
181]. Nonetheless, targeting of the proteolytic machinery is primarily oriented to proteasome inhibition, with three inhibitors—bortezomib, carfilzomib and ixazomib—already approved for the treatment of multiple myeloma and mantle cell lymphoma [
182], while other agents, such as delanzomib (CEP-18770) and marizomib or salinosporamide A (NPI-0052) [
183] are currently being tested in clinical trials as potential treatment options for solid tumors (NCT00572637 and NCT03345095, respectively).
7. The WNT/b Catenin Pathway
The WNT/b catenin pathway is one of the best-studied signaling cascades in cancer development, known to be implicated in several cancer-related cellular functions, such as cell proliferation, stem cell maintenance, differentiation, cell–cell adhesion, morphogenetic processes, migration, angiogenesis and immune evasion [
184,
185,
186,
187,
188]. Mutations in WNT/b catenin components are very common in cancer patients. Our analysis revealed that carcinogenic mutational occurrences in genes of this pathway are limited compared to the pathways discussed so far. Specifically, the proportion of patients with at least one driver mutation in this pathway exceeded 10% in only nine of the thirty-six cancer types examined here. Intestinal malignancies ranked highest in terms of mutational landscape with at least 80% of colon adenocarcinoma, rectal adenocarcinoma and mucinous adenocarcinoma of the colon and rectum patients harboring driver alterations. On the other hand, no such mutations were found in uveal melanoma or leiomyosarcoma patients (
Figure 1). Among WNT/b catenin effectors, the
APC,
CTNNB1 and
RNF43 genes were found mutated in 48.59%, 26.41% and 14.94% of the affected samples, respectively, while four other genes (in descending order of mutational frequency:
AMER1,
AXIN1,
TCF7L2 and
AXIN2) exhibited a driver mutational rate between ~5% and ~8%.
The most frequent mutations are nonsense mutations in the
APC tumor suppressor, present in 40.6% of patients. Conversion of arginine-encoding codons into stop codons predominantly takes place at positions 1450, 876 and 1114 of the APC protein (8.51%, 4.84% and 4.64% respectively). R1450 is located into the so-called mutation cluster region (MCR) [
189]. Truncation of the APC protein in this position leads to loss of all axin- and most b catenin-binding sites, therefore abrogating the ability of APC to negatively regulate b catenin via the formation of the destruction complex AXIN-APC-CK1alpha-GSK3beta [
190,
191,
192,
193]. Similarly, two other truncations, namely the R1114* and R876* mutants, also lose the axin- and most b catenin-binding sites [
194,
195]. In all three cases, the subsequent b catenin accumulation and nuclear entry permits activation of the TCF/LEF1 transcriptional complex, thereby promoting cellular proliferation and tumorigenesis [
192].
Somatic mutations in the
CTNNB1 (catenin-beta 1) gene, which is involved in the regulation of cell adhesion and gene transcription, are almost exclusively missense mutations. In particular, aa substitutions at six specific positions (32, 33, 34, 37, 41 and 45) represent more than 85% of all affected patients in our dataset. Among them, S33, S37 and D32 replacements were the most prevalent, affecting 18.51%, 17.44% and 16.01% of all cases, respectively. The protein region between D32 and S45 participates in the phosphorylation of b catenin from CK1alpha and GSK3beta (components of its destruction complex), as well as in the interaction of phosphorylated b catenin with its E3 ligase substrate recognition component, FBW1 [
196,
197]. Consequently, mutations in these protein sites exert similar signaling implications to the ones exerted by APC, as b catenin escapes proteasomal degradation and confers an increased proliferative potential to the cell [
197,
198,
199,
200,
201].
Another gene found mutated in cancer patients is the
RNF43 (Ring Finger Protein 43), a downstream target gene of Wnt/b catenin signaling. Almost seventy-three percent of
RNF43-mutated primary tumors carry a frameshift deletion in this gene, while the second most frequent alteration type is a non-sense mutation (~17%). By far the most common alteration of the RNF43 protein is G659Vfs*41, being present in approximately 60% of all
RNF43-mutated tumors. Interestingly, despite its recurrent presence in cancer samples, this frameshift deletion leaves protein function intact, with the relevant RNF43-mutant being able to exert its E3 ubiquitin ligase activity [
202]; this results in the tagging of FZD family (frizzled transmembrane proteins) WNT receptors for proteasomal degradation and in the inactivation of WNT/b catenin signaling [
203,
204]. The second and third most frequent somatic mutations of the RNF43 protein are R519* and R145*, affecting only 2.52% and 1.89% of all carriers, respectively. Both of these aberrations lead to a truncated protein with a WNT signaling-enhancing role, however the exact mechanism differs depending on the truncation position. The catalytic RING domain of RNF43 lies among P270 and I316 [
202,
205], and as such the R519* mutant retains its E3 ubiquitin ligase activity. Nonetheless, it simultaneously gains the ability to snare CK1alpha at the plasma membrane, thus assisting b catenin to escape degradation [
206]. On the other hand, R145* variants lack this catalytic activity and are therefore expected to abort their FZD degradation and b catenin destabilization role [
202].
Significant efforts have been made to therapeutically target the WNT/b catenin network, mostly for the development of small molecule stabilizers of the b catenin destruction complex components or destabilizers of the b catenin-TCF/LEF interaction, but also for the development of antibodies and regulatory peptides that directly or indirectly affect—mostly inhibit—WNT or FZD proteins [
192,
207,
208]. Four such constructs have exhibited the most encouraging results and their efficacy is currently evaluated in phase II clinical trials of both solid and blood cancer patients. In this context, WNT974 (NCT02278133)—a porcupine inhibitor that impedes WNT secretion and activity, Foxy-5 (NCT03883802)—a WNT5a mimetic, PRI-724 (NCT01606579)—an antagonist of the b catenin coactivator CBP, and DKN-01 (NCT03395080)—a monoclonal antibody that neutralizes the activity of WNT/b catenin axis inhibitor DKK1, are now in the spotlight of WNT pathway targeting.
8. The Notch Pathway
Notch is a cancer-related signaling pathway well-known for its involvement in a variety of developmental processes, as well as cellular differentiation, proliferation, stem cell maintenance, angiogenesis, EMT, inflammation and apoptosis [
209,
210,
211]. Mutations in the Notch pathway, were found in at least one in ten patients in fifteen out of thirty-six assessed cancer types. Uterine carcinosarcoma exhibited the highest driver mutational rate (40.35%), followed by mucinous adenocarcinoma of the colon and rectum (33.93%) and bladder urothelial carcinoma (29.02%), while other gynecological and upper digestive tract malignancies, were also highly affected. On the other hand, none of the patients with pheochromocytoma and no more than 2% of patients with leiomyosarcoma, uveal melanoma and papillary thyroid cancer carried such mutations (
Figure 1). At the gene level, the most frequently mutated components or regulators of the Notch pathway appeared to be identical to the above discussed ubiquitination pathways;
FBXW7,
EP300 and
CREBBP were found altered in 33.65%, 18.41% and 16.97% of Notch-impaired tumors, respectively, while somatic driver events in each of the
SPEN,
NCOR1 and
NOTCH1 genes, were detected in ~12% of cases. Upon activation of the NOTCH receptors by their ligands (e.g., delta-like protein 1, protein jagged-1 etc), the NOTCH-intracellular domain (NICD) is released and transferred into the nucleus where it acts as a transcription regulator. NICD is one of the FBW7 substrates [
159] and mutations within the FBW7 WD40 domain have been reported to impede this interplay, therefore leading to NICD accumulation and enhanced Notch signaling, which may lead to a tumorigenic outcome [
157,
212,
213]. Furthermore, despite the previously reported p300 requirement for NICD transcriptional activity [
214,
215], it was recently demonstrated that loss-of-function mutations in either the
EP300 or
CREBBP gene can also activate the Notch axis, due to the subsequent low histone acetylation levels of the
FBXW7 promoter [
216], suggesting the implication of additional transcriptional NICD co-activators.
So far, no targeted therapies for Notch signaling regulation have entered the clinical practice. However, remarkable efforts have been made to overcome the obstacles associated with Notch pathway signaling, given the highly context-specific behavior of this signaling axis in cancer [
217]. To this end, diverse strategies have been utilized, such as the targeting of NOTCH biosynthesis enzymes, receptor-ligand interplay, NOTCH cleavage-performing effectors or NICD-containing transcriptional complexes assemblage [
217,
218,
219]. The most encouraging results come from the development of inhibitors against gamma-secretase—which is responsible for the final NICD-releasing NOTCH cleavage—and delta-like protein 3, a NOTCH ligand, with some of the relevant cancer-related clinical trials being in advanced stages. Specifically, the efficacy of gamma-secretase inhibitors (GSIs) nirogacestat/PF-03084014 (NCT03785964) and MK0752 (NCT00756717) is currently evaluated in phase III trials in adults with desmoid tumor and in early stage breast cancer patients in combination with tamoxifen respectively, while the tesirine conjugated anti-delta-like protein 3 mAb rovalpituzumab (Rova-T) (NCT03061812) is being tested in a phase III clinical trial in small-cell lung cancer (SCLC) patients with disease progression following platinum-based chemotherapy and overexpressing delta-like protein 3.
This entry is adapted from the peer-reviewed paper 10.3390/cancers14030664