 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Francisco Nualart | -- | 3195 | 2022-06-08 23:10:58 | | | |
| 2 | Vivi Li | -32 word(s) | 3163 | 2022-06-09 06:01:56 | | |
Video Upload Options
Historically, vitamin C has been associated with many regulatory processes that involve specific signaling pathways. Among the most studied signaling pathways are those involved in the regulation of aging, differentiation, neurotransmission, proliferation, and cell death processes in cancer. This wide variety of regulatory effects is due to the fact that vitamin C has a dual mechanism of action. The reduced form of vitamin C (ascorbic acid, AA) is an essential micronutrient of small size; it is soluble in water and has two dissociable protons with pKa values of 4.2 and 11.8. At physiological pH, its reduced form predominates as the monovalent ascorbate anion (AA); when it loses the second proton, it is oxidized to dehydroascorbic acid (DHA).
1. Introduction
2. Molecular Pathways Regulated by Vitamin C
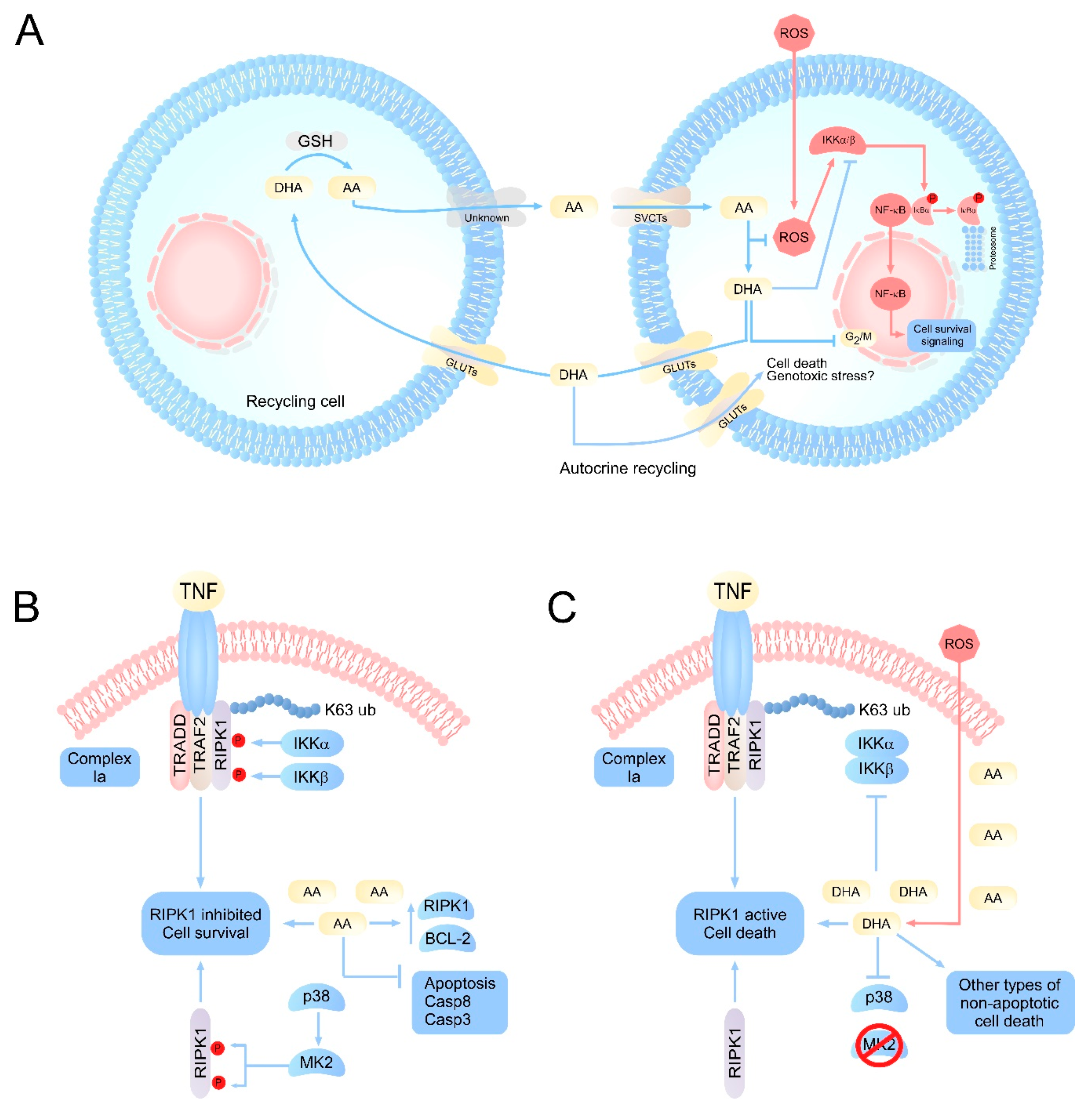
3. Vitamin C as a Cell Cycle Regulator
4. Vitamin C as an Enzymatic Cofactor
5. Regulation of Necroptosis by Vitamin C: AA as Apoptosis Inhibitor and DHA as RIPK1 Activator
References
- Padayatty, S.J.; Levine, M. Vitamin C: The known and the unknown and Goldilocks. Oral Dis. 2016, 22, 463–493.
- May, J.M. Vitamin C transport and its role in the central nervous system. Subcell. Biochem. 2012, 56, 85–103.
- Nualart, F.; Mack, L.; Garcia, A.; Cisternas, P.; Bongarzone, E.R.; Heitzer, M.; Jara, N.; Martinez, F.; Ferrada, L.; Espinoza, F.; et al. Vitamin C Transporters, Recycling and the Bystander Effect in the Nervous System: SVCT2 versus Gluts. J. Stem Cell Res. Ther. 2014, 4, 209.
- Nishikimi, M.; Fukuyama, R.; Minoshima, S.; Shimizu, N.; Yagi, K. Cloning and chromosomal mapping of the human nonfunctional gene for L-gulono-gamma-lactone oxidase, the enzyme for L-ascorbic acid biosynthesis missing in man. J. Biol. Chem. 1994, 269, 13685–13688.
- Harrison, F.E.; May, J.M. Vitamin C function in the brain: Vital role of the ascorbate transporter SVCT2. Free Radic. Biol. Med. 2009, 46, 719–730.
- Rice, M.E. Ascorbate regulation and its neuroprotective role in the brain. Trends Neurosci. 2000, 23, 209–216.
- Ngo, B.; Van Riper, J.M.; Cantley, L.C.; Yun, J. Targeting cancer vulnerabilities with high-dose vitamin C. Nat. Rev. Cancer 2019, 19, 271–282.
- Tsukaguchi, H.; Tokui, T.; Mackenzie, B.; Berger, U.V.; Chen, X.Z.; Wang, Y.; Brubaker, R.F.; Hediger, M.A. A family of mammalian Na+-dependent L-ascorbic acid transporters. Nature 1999, 399, 70–75.
- Silva-Alvarez, C.; Salazar, K.; Cisternas, P.; Martinez, F.; Liour, S.; Jara, N.; Bertinat, R.; Nualart, F. Apical Polarization of SVCT2 in Apical Radial Glial Cells and Progenitors During Brain Development. Mol. Neurobiol. 2017, 54, 5449–5467.
- Salazar, K.; Martinez, M.; Ulloa, V.; Bertinat, R.; Martinez, F.; Jara, N.; Espinoza, F.; Bongarzone, E.R.; Nualart, F. SVCT2 Overexpression in Neuroblastoma Cells Induces Cellular Branching that is Associated with ERK Signaling. Mol. Neurobiol. 2016, 53, 6668–6679.
- Wang, Y.; Mackenzie, B.; Tsukaguchi, H.; Weremowicz, S.; Morton, C.C.; Hediger, M.A. Human vitamin C (L-ascorbic acid) transporter SVCT1. Biochem. Biophys. Res. Commun. 2000, 267, 488–494.
- Ferrada, L.; Salazar, K.; Nualart, F. Metabolic control by dehydroascorbic acid: Questions and controversies in cancer cells. J. Cell Physiol. 2019, 234, 19331–19338.
- Nualart, F.J.; Rivas, C.I.; Montecinos, V.P.; Godoy, A.S.; Guaiquil, V.H.; Golde, D.W.; Vera, J.C. Recycling of vitamin C by a bystander effect. J. Biol. Chem. 2003, 278, 10128–10133.
- Rumsey, S.C.; Daruwala, R.; Al-Hasani, H.; Zarnowski, M.J.; Simpson, I.A.; Levine, M. Dehydroascorbic acid transport by GLUT4 in Xenopus oocytes and isolated rat adipocytes. J. Biol. Chem. 2000, 275, 28246–28253.
- Rumsey, S.C.; Kwon, O.; Xu, G.W.; Burant, C.F.; Simpson, I.; Levine, M. Glucose transporter isoforms GLUT1 and GLUT3 transport dehydroascorbic acid. J. Biol. Chem. 1997, 272, 18982–18989.
- Corpe, C.P.; Eck, P.; Wang, J.; Al-Hasani, H.; Levine, M. Intestinal dehydroascorbic acid (DHA) transport mediated by the facilitative sugar transporters, GLUT2 and GLUT8. J. Biol. Chem. 2013, 288, 9092–9101.
- Cisternas, P.; Silva-Alvarez, C.; Martinez, F.; Fernandez, E.; Ferrada, L.; Oyarce, K.; Salazar, K.; Bolanos, J.P.; Nualart, F. The oxidized form of vitamin C, dehydroascorbic acid, regulates neuronal energy metabolism. J. Neurochem. 2014, 129, 663–671.
- Garcia-Krauss, A.; Ferrada, L.; Astuya, A.; Salazar, K.; Cisternas, P.; Martinez, F.; Ramirez, E.; Nualart, F. Dehydroascorbic Acid Promotes Cell Death in Neurons Under Oxidative Stress: A Protective Role for Astrocytes. Mol. Neurobiol. 2016, 53, 5847–5863.
- Ferrada, L.; Barahona, M.J.; Salazar, K.; Vandenabeele, P.; Nualart, F. Vitamin C controls neuronal necroptosis under oxidative stress. Redox Biol. 2020, 29, 101408.
- Bowie, A.G.; O’Neill, L.A. Vitamin C inhibits NF-kappa B activation by TNF via the activation of p38 mitogen-activated protein kinase. J. Immunol. 2000, 165, 7180–7188.
- Bowie, A.; O’Neill, L.A. Vitamin C inhibits NF kappa B activation in endothelial cells. Biochem. Soc. Trans. 1997, 25, 131S.
- Carcamo, J.M.; Pedraza, A.; Borquez-Ojeda, O.; Golde, D.W. Vitamin C suppresses TNF alpha-induced NF kappa B activation by inhibiting I kappa B alpha phosphorylation. Biochemistry 2002, 41, 12995–13002.
- Abhilash, P.A.; Harikrishnan, R.; Indira, M. Ascorbic acid suppresses endotoxemia and NF-kappaB signaling cascade in alcoholic liver fibrosis in guinea pigs: A mechanistic approach. Toxicol. Appl. Pharmacol. 2014, 274, 215–224.
- Yi, F.; Frazzette, N.; Cruz, A.C.; Klebanoff, C.A.; Siegel, R.M. Beyond Cell Death: New Functions for TNF Family Cytokines in Autoimmunity and Tumor Immunotherapy. Trends Mol. Med. 2018, 24, 642–653.
- Malek, R.; Borowicz, K.K.; Jargiello, M.; Czuczwar, S.J. Role of nuclear factor kappaB in the central nervous system. Pharmacol. Rep. 2007, 59, 25–33.
- Engelmann, C.; Weih, F.; Haenold, R. Role of nuclear factor kappa B in central nervous system regeneration. Neural Regen. Res. 2014, 9, 707–711.
- Carcamo, J.M.; Pedraza, A.; Borquez-Ojeda, O.; Zhang, B.; Sanchez, R.; Golde, D.W. Vitamin C is a kinase inhibitor: Dehydroascorbic acid inhibits IkappaBalpha kinase beta. Mol. Cell. Biol. 2004, 24, 6645–6652.
- Lee, S.A.; Son, Y.O.; Kook, S.H.; Choi, K.C.; Lee, J.C. Ascorbic acid increases the activity and synthesis of tyrosinase in B16F10 cells through activation of p38 mitogen-activated protein kinase. Arch. Dermatol. Res. 2011, 303, 669–678.
- Lei, F.X.; Jin, L.; Liu, X.Y.; Lai, F.; Yan, X.G.; Farrelly, M.; Guo, S.T.; Zhao, X.H.; Zhang, X.D. RIP1 protects melanoma cells from apoptosis induced by BRAF/MEK inhibitors. Cell Death Dis. 2018, 9, 679.
- Park, S.; Park, C.H.; Hahm, E.R.; Kim, K.; Kimler, B.F.; Lee, S.J.; Park, H.K.; Lee, S.H.; Kim, W.S.; Jung, C.W.; et al. Activation of Raf1 and the ERK pathway in response to l-ascorbic acid in acute myeloid leukemia cells. Cell Signal. 2005, 17, 111–119.
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Pooput, C.; Kirk, K.L.; Krishna, M.C.; Khosh, D.B.; Drisko, J.; Levine, M. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 11105–11109.
- Chen, Q.; Espey, M.G.; Krishna, M.C.; Mitchell, J.B.; Corpe, C.P.; Buettner, G.R.; Shacter, E.; Levine, M. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: Action as a pro-drug to deliver hydrogen peroxide to tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 13604–13609.
- Heaney, M.L.; Gardner, J.R.; Karasavvas, N.; Golde, D.W.; Scheinberg, D.A.; Smith, E.A.; O’Connor, O.A. Vitamin C antagonizes the cytotoxic effects of antineoplastic drugs. Cancer Res. 2008, 68, 8031–8038.
- Perez-Cruz, I.; Carcamo, J.M.; Golde, D.W. Vitamin C inhibits FAS-induced apoptosis in monocytes and U937 cells. Blood 2003, 102, 336–343.
- Perez-Cruz, I.; Carcamo, J.M.; Golde, D.W. Caspase-8 dependent TRAIL-induced apoptosis in cancer cell lines is inhibited by vitamin C and catalase. Apoptosis 2007, 12, 225–234.
- Zinkel, S.; Gross, A.; Yang, E. BCL2 family in DNA damage and cell cycle control. Cell Death Differ. 2006, 13, 1351–1359.
- Nguyen, M.D.; Mushynski, W.E.; Julien, J.P. Cycling at the interface between neurodevelopment and neurodegeneration. Cell Death Differ. 2002, 9, 1294–1306.
- El Banna, N.; Hatem, E.; Heneman-Masurel, A.; Leger, T.; Baille, D.; Vernis, L.; Garcia, C.; Martineau, S.; Dupuy, C.; Vagner, S.; et al. Redox modifications of cysteine-containing proteins, cell cycle arrest and translation inhibition: Involvement in vitamin C-induced breast cancer cell death. Redox. Biol. 2019, 26, 101290.
- Belin, S.; Kaya, F.; Duisit, G.; Giacometti, S.; Ciccolini, J.; Fontes, M. Antiproliferative effect of ascorbic acid is associated with the inhibition of genes necessary to cell cycle progression. PLoS ONE 2009, 4, e4409.
- Martinotti, S.; Ranzato, E.; Parodi, M.; Vitale, M.; Burlando, B. Combination of ascorbate/epigallocatechin-3-gallate/gemcitabine synergistically induces cell cycle deregulation and apoptosis in mesothelioma cells. Toxicol. Appl. Pharmacol. 2014, 274, 35–41.
- Felipe, K.B.; Benites, J.; Glorieux, C.; Sid, B.; Valenzuela, M.; Kviecinski, M.R.; Pedrosa, R.C.; Valderrama, J.A.; Leveque, P.; Gallez, B.; et al. Antiproliferative effects of phenylaminonaphthoquinones are increased by ascorbate and associated with the appearance of a senescent phenotype in human bladder cancer cells. Biochem. Biophys. Res. Commun. 2013, 433, 573–578.
- McConnell, M.J.; Herst, P.M. Ascorbate combination therapy: New tool in the anticancer toolbox? Sci. Transl. Med. 2014, 6, 222fs6.
- Alexander, B.; Fishman, A.I.; Eshghi, M.; Choudhury, M.; Konno, S. Induction of cell death in renal cell carcinoma with combination of D-fraction and vitamin C. Integr. Cancer Ther. 2013, 12, 442–448.
- Herst, P.M.; Broadley, K.W.; Harper, J.L.; McConnell, M.J. Pharmacological concentrations of ascorbate radiosensitize glioblastoma multiforme primary cells by increasing oxidative DNA damage and inhibiting G2/M arrest. Free. Radic. Biol. Med. 2012, 52, 1486–1493.
- Moteki, H.; Shimamura, Y.; Kimura, M.; Ogihara, M. Signal transduction pathway for L-ascorbic acid- and L-ascorbic acid 2-glucoside-induced DNA synthesis and cell proliferation in primary cultures of adult rat hepatocytes. Eur. J. Pharmacol. 2012, 683, 276–284.
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation 2011, 123, 2145–2156.
- Lu, C.; Thompson, C.B. Metabolic regulation of epigenetics. Cell Metab. 2012, 16, 9–17.
- Huang, S.; Xu, Y.M.; Lau, A.T.Y. Epigenetic effects of the 13 vitamins. Curr. Pharmacol. Rep. 2018, 4, 453–467.
- Kuiper, C.; Vissers, M.C. Ascorbate as a co-factor for fe- and 2-oxoglutarate dependent dioxygenases: Physiological activity in tumor growth and progression. Front. Oncol. 2014, 4, 359.
- Melamed, P.; Yosefzon, Y.; David, C.; Tsukerman, A.; Pnueli, L. Tet Enzymes, Variants, and Differential Effects on Function. Front. Cell Dev. Biol. 2018, 6, 22.
- Carr, A.C.; Frei, B. Toward a new recommended dietary allowance for vitamin C based on antioxidant and health effects in humans. Am. J. Clin. Nutr. 1999, 69, 1086–1107.
- Farrow, S.C.; Facchini, P.J. Functional diversity of 2-oxoglutarate/Fe(II)-dependent dioxygenases in plant metabolism. Front. Plant. Sci. 2014, 5, 524.
- Myllyla, R.; Majamaa, K.; Gunzler, V.; Hanauske-Abel, H.M.; Kivirikko, K.I. Ascorbate is consumed stoichiometrically in the uncoupled reactions catalyzed by prolyl 4-hydroxylase and lysyl hydroxylase. J. Biol. Chem. 1984, 259, 5403–5405.
- de Jong, L.; Albracht, S.P.; Kemp, A. Prolyl 4-hydroxylase activity in relation to the oxidation state of enzyme-bound iron. The role of ascorbate in peptidyl proline hydroxylation. Biochim. Biophys. Acta 1982, 704, 326–332.
- Cimmino, L.; Neel, B.G.; Aifantis, I. Vitamin C in Stem Cell Reprogramming and Cancer. Trends Cell Biol. 2018, 28, 698–708.
- Lane, D.J.; Chikhani, S.; Richardson, V.; Richardson, D.R. Transferrin iron uptake is stimulated by ascorbate via an intracellular reductive mechanism. Biochim. Biophys. Acta 2013, 1833, 1527–1541.
- Tsukada, Y.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.H.; Tempst, P.; Zhang, Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2006, 439, 811–816.
- Yin, R.; Mao, S.Q.; Zhao, B.; Chong, Z.; Yang, Y.; Zhao, C.; Zhang, D.; Huang, H.; Gao, J.; Li, Z.; et al. Ascorbic acid enhances Tet-mediated 5-methylcytosine oxidation and promotes DNA demethylation in mammals. J. Am. Chem. Soc. 2013, 135, 10396–10403.
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123.
- Zhang, D.W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.J.; Lin, S.C.; Dong, M.Q.; Han, J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009, 325, 332–336.
- He, S.; Wang, L.; Miao, L.; Wang, T.; Du, F.; Zhao, L.; Wang, X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009, 137, 1100–1111.
- Wang, L.; Du, F.; Wang, X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008, 133, 693–703.
- Pasparakis, M.; Vandenabeele, P. Necroptosis and its role in inflammation. Nature 2015, 517, 311–320.
- Vanden Berghe, T.; Kaiser, W.J.; Bertrand, M.J.; Vandenabeele, P. Molecular crosstalk between apoptosis, necroptosis, and survival signaling. Mol. Cell. Oncol. 2015, 2, e975093.
- Murphy, J.M.; Czabotar, P.E.; Hildebrand, J.M.; Lucet, I.S.; Zhang, J.G.; Alvarez-Diaz, S.; Lewis, R.; Lalaoui, N.; Metcalf, D.; Webb, A.I.; et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 2013, 39, 443–453.
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227.
- Hildebrand, J.M.; Tanzer, M.C.; Lucet, I.S.; Young, S.N.; Spall, S.K.; Sharma, P.; Pierotti, C.; Garnier, J.M.; Dobson, R.C.; Webb, A.I.; et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc. Natl. Acad. Sci. USA 2014, 111, 15072–15077.
- Zhang, Y.; Chen, X.; Gueydan, C.; Han, J. Plasma membrane changes during programmed cell deaths. Cell Res. 2018, 28, 9–21.
- Meng, H.; Liu, Z.; Li, X.; Wang, H.; Jin, T.; Wu, G.; Shan, B.; Christofferson, D.E.; Qi, C.; Yu, Q.; et al. Death-domain dimerization-mediated activation of RIPK1 controls necroptosis and RIPK1-dependent apoptosis. Proc. Natl. Acad. Sci. USA 2018, 115, E2001–E2009.
- Amin, P.; Florez, M.; Najafov, A.; Pan, H.; Geng, J.; Ofengeim, D.; Dziedzic, S.A.; Wang, H.; Barrett, V.J.; Ito, Y.; et al. Regulation of a distinct activated RIPK1 intermediate bridging complex I and complex II in TNFalpha-mediated apoptosis. Proc. Natl. Acad. Sci. USA 2018.
- Remijsen, Q.; Goossens, V.; Grootjans, S.; Van den Haute, C.; Vanlangenakker, N.; Dondelinger, Y.; Roelandt, R.; Bruggeman, I.; Goncalves, A.; Bertrand, M.J.; et al. Depletion of RIPK3 or MLKL blocks TNF-driven necroptosis and switches towards a delayed RIPK1 kinase-dependent apoptosis. Cell Death Dis. 2014, 5, e1004.
- Degterev, A.; Yuan, J. Expansion and evolution of cell death programmes. Nat. Rev. Mol. Cell Biol. 2008, 9, 378–390.
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’en, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321.
- de Almagro, M.C.; Goncharov, T.; Izrael-Tomasevic, A.; Duttler, S.; Kist, M.; Varfolomeev, E.; Wu, X.; Lee, W.P.; Murray, J.; Webster, J.D.; et al. Coordinated ubiquitination and phosphorylation of RIP1 regulates necroptotic cell death. Cell Death Differ. 2017, 24, 26–37.
- Bertrand, M.J.; Milutinovic, S.; Dickson, K.M.; Ho, W.C.; Boudreault, A.; Durkin, J.; Gillard, J.W.; Jaquith, J.B.; Morris, S.J.; Barker, P.A. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 2008, 30, 689–700.
- Dondelinger, Y.; Jouan-Lanhouet, S.; Divert, T.; Theatre, E.; Bertin, J.; Gough, P.J.; Giansanti, P.; Heck, A.J.; Dejardin, E.; Vandenabeele, P.; et al. NF-kappaB-Independent Role of IKKalpha/IKKbeta in Preventing RIPK1 Kinase-Dependent Apoptotic and Necroptotic Cell Death during TNF Signaling. Mol. Cell 2015, 60, 63–76.
- Dondelinger, Y.; Delanghe, T.; Rojas-Rivera, D.; Priem, D.; Delvaeye, T.; Bruggeman, I.; Van Herreweghe, F.; Vandenabeele, P.; Bertrand, M.J.M. MK2 phosphorylation of RIPK1 regulates TNF-mediated cell death. Nat. Cell Biol. 2017, 19, 1237–1247.
- Jaco, I.; Annibaldi, A.; Lalaoui, N.; Wilson, R.; Tenev, T.; Laurien, L.; Kim, C.; Jamal, K.; Wicky John, S.; Liccardi, G.; et al. MK2 Phosphorylates RIPK1 to Prevent TNF-Induced Cell Death. Mol. Cell 2017, 66, 698–710.e695.
- Shan, B.; Pan, H.; Najafov, A.; Yuan, J. Necroptosis in development and diseases. Genes Dev. 2018, 32, 327–340.
- Christofferson, D.E.; Li, Y.; Yuan, J. Control of life-or-death decisions by RIP1 kinase. Annu. Rev. Physiol. 2014, 76, 129–150.
- Dhar-Mascareno, M.; Carcamo, J.M.; Golde, D.W. Hypoxia-reoxygenation-induced mitochondrial damage and apoptosis in human endothelial cells are inhibited by vitamin C. Free. Radic. Biol. Med. 2005, 38, 1311–1322.
- Guaiquil, V.H.; Golde, D.W.; Beckles, D.L.; Mascareno, E.J.; Siddiqui, M.A. Vitamin C inhibits hypoxia-induced damage and apoptotic signaling pathways in cardiomyocytes and ischemic hearts. Free. Radic. Biol. Med. 2004, 37, 1419–1429.
- Han, J.M.; Chang, B.J.; Li, T.Z.; Choe, N.H.; Quan, F.S.; Jang, B.J.; Cho, I.H.; Hong, H.N.; Lee, J.H. Protective effects of ascorbic acid against lead-induced apoptotic neurodegeneration in the developing rat hippocampus in vivo. Brain Res. 2007, 1185, 68–74.
- Tian, W.; Wang, Y.; Xu, Y.; Guo, X.; Wang, B.; Sun, L.; Liu, L.; Cui, F.; Zhuang, Q.; Bao, X.; et al. The hypoxia-inducible factor renders cancer cells more sensitive to vitamin C-induced toxicity. J. Biol. Chem. 2014, 289, 3339–3351.
- Evans, M.K.; Tovmasyan, A.; Batinic-Haberle, I.; Devi, G.R. Mn porphyrin in combination with ascorbate acts as a pro-oxidant and mediates caspase-independent cancer cell death. Free Radic. Biol. Med. 2014, 68, 302–314.
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.C.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396.
- Sarosiek, K.A.; Fraser, C.; Muthalagu, N.; Bhola, P.D.; Chang, W.; McBrayer, S.K.; Cantlon, A.; Fisch, S.; Golomb-Mello, G.; Ryan, J.A.; et al. Developmental Regulation of Mitochondrial Apoptosis by c-Myc Governs Age- and Tissue-Specific Sensitivity to Cancer Therapeutics. Cancer Cell 2017, 31, 142–156.
- Kole, A.J.; Annis, R.P.; Deshmukh, M. Mature neurons: Equipped for survival. Cell Death Dis. 2013, 4, e689.



