 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mark R Woodford | -- | 2637 | 2022-06-07 17:02:29 | | | |
| 2 | Peter Tang | Meta information modification | 2637 | 2022-06-08 07:46:19 | | |
Video Upload Options
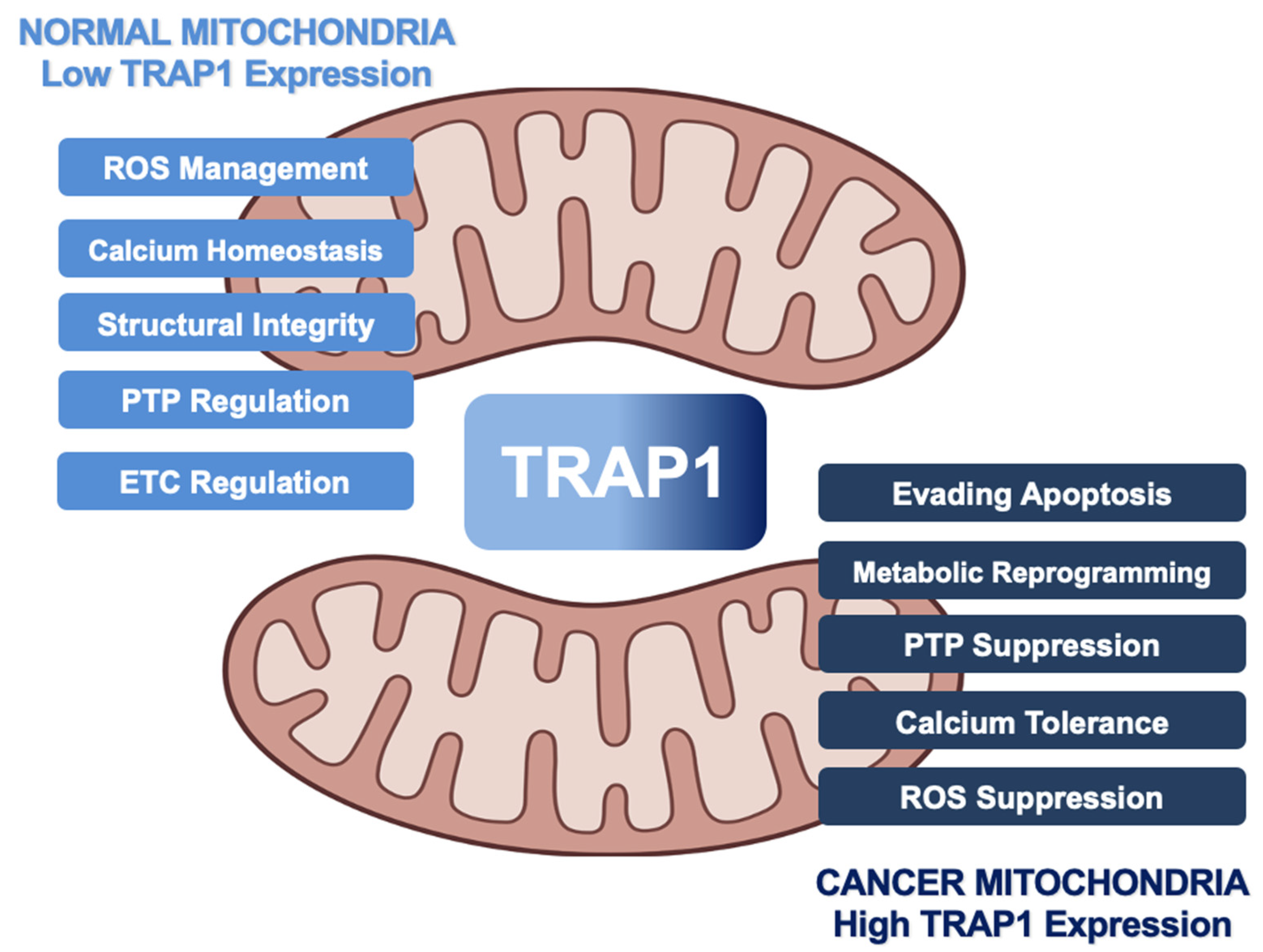
The Hsp90 chaperone TNF-receptor-associated protein-1 (TRAP1) is primarily localized to the mitochondria and controls both cellular metabolic reprogramming and mitochondrial apoptosis. TRAP1 upregulation facilitates the growth and progression of many cancers by promoting glycolytic metabolism and antagonizing the mitochondrial permeability transition that precedes multiple cell death pathways. TRAP1 attenuation induces apoptosis in cellular models of cancer, identifying TRAP1 as a potential therapeutic target in cancer. Similar to cytosolic Hsp90 proteins, TRAP1 is also subject to post-translational modifications (PTM) that regulate its function and mediate its impact on downstream effectors, or ‘clients’.
1. Introduction
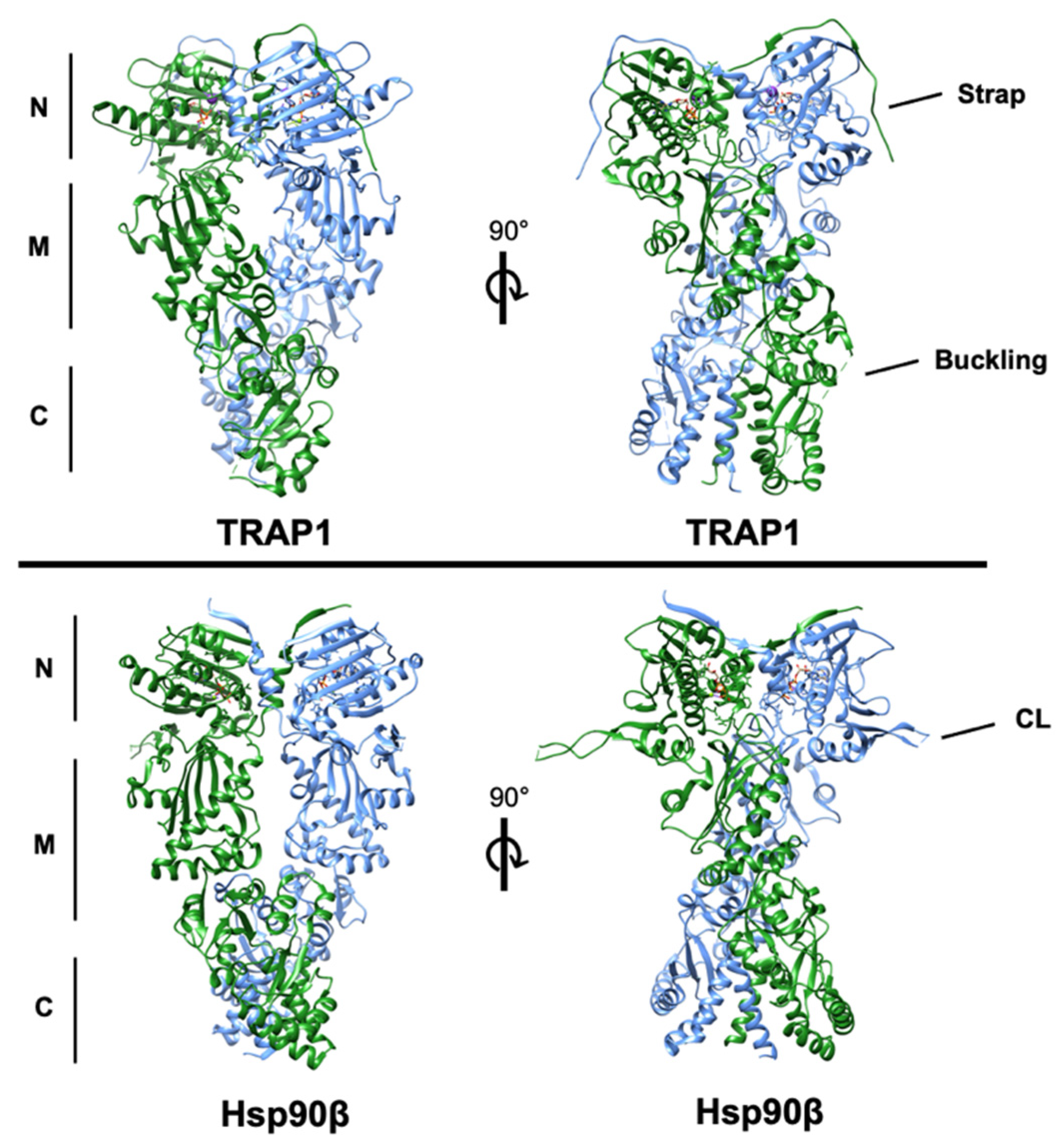
2. Structural Basis of TRAP1 Activity
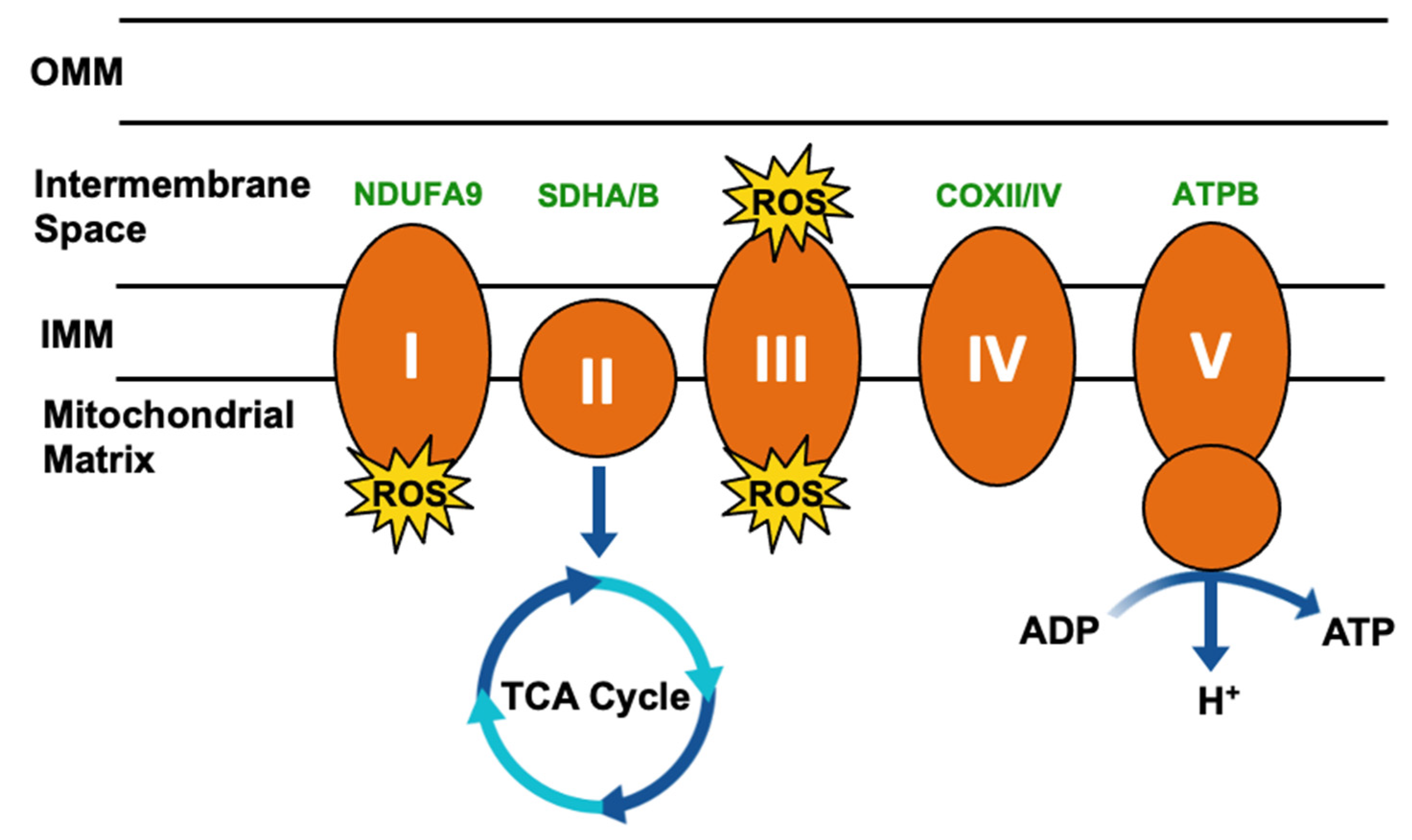
3. Impact of TRAP1 on Cancer Metabolism
3.1. Metabolic Regulation
3.2. Contribution to Tumorigenesis
3.3. Evasion of Apoptosis
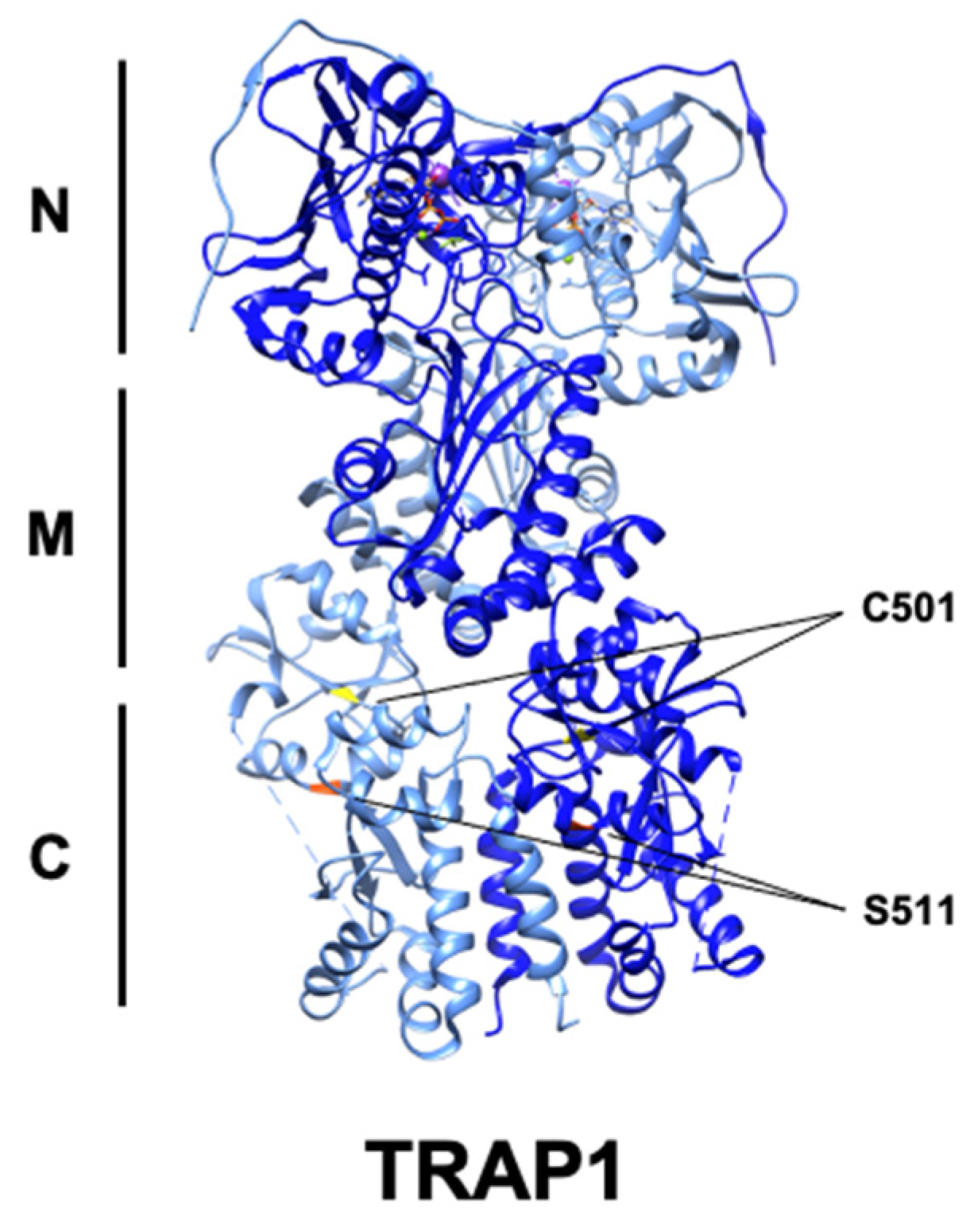
4. Post-Translational Regulation of TRAP1
|
Modification |
Enzyme |
Residue |
Paralog |
Impact on TRAP1 |
Reference |
|---|---|---|---|---|---|
|
S-Nitrosylation |
GSNOR |
Cys501 |
Thr495 |
Decreased activity, proteasomal degradation |
[98] |
|
Phosphorylation |
ERK1/2 |
Ser511 |
Ser505 |
N/A |
[10] |
|
Phosphorylation |
ERK1/2 |
Ser568 |
Glu562 |
Increased SDH inhibition |
[10] |
|
S/T Phosphorylation |
PINK1 |
N/A |
N/A |
N/A |
[5] |
|
Y Phosphorylation |
Unknown, possibly c-Src |
N/A |
N/A |
Disrupts c-Src interaction |
[6] |
|
Deacetylation |
SIRT3 |
N/A |
N/A |
Increased activity |
[27] |
References
- Prodromou, C.; Bjorklund, D.M. Advances towards Understanding the Mechanism of Action of the Hsp90 Complex. Biomolecules 2022, 12, 600.
- Johnson, J.L. Mutations in Hsp90 Cochaperones Result in a Wide Variety of Human Disorders. Front. Mol. Biosci. 2021, 8, 787260.
- Backe, S.J.; Sager, R.A.; Woodford, M.R.; Makedon, A.M.; Mollapour, M. Post-Translational Modifications of Hsp90 and Translating the Chaperone Code. J. Biol. Chem. 2020, 295, 11099–11117.
- Cechetto, J.D.; Gupta, R.S. Immunoelectron Microscopy Provides Evidence That Tumor Necrosis Factor Receptor-Associated Protein 1 (TRAP-1) Is a Mitochondrial Protein Which Also Localizes at Specific Extramitochondrial Sites. Exp. Cell Res. 2000, 260, 30–39.
- Pridgeon, J.W.; Olzmann, J.A.; Chin, L.-S.; Li, L. PINK1 Protects against Oxidative Stress by Phosphorylating Mitochondrial Chaperone TRAP1. PLoS Biol. 2007, 5, e172.
- Yoshida, S.; Tsutsumi, S.; Muhlebach, G.; Sourbier, C.; Lee, M.-J.; Lee, S.; Vartholomaiou, E.; Tatokoro, M.; Beebe, K.; Miyajima, N.; et al. Molecular Chaperone TRAP1 Regulates a Metabolic Switch between Mitochondrial Respiration and Aerobic Glycolysis. Proc. Natl. Acad. Sci. USA 2013, 110, E1604–E1612.
- Song, H.Y.; Dunbar, J.D.; Zhang, Y.X.; Guo, D.; Donner, D.B. Identification of a Protein with Homology to Hsp90 That Binds the Type 1 Tumor Necrosis Factor Receptor. J. Biol. Chem. 1995, 270, 3574–3581.
- Felts, S.J.; Owen, B.A.L.; Nguyen, P.; Trepel, J.; Donner, D.B.; Toft, D.O. The Hsp90-Related Protein TRAP1 Is a Mitochondrial Protein with Distinct Functional Properties. J. Biol. Chem. 2000, 275, 3305–3312.
- Cannino, G.; Ciscato, F.; Masgras, I.; Sánchez-Martín, C.; Rasola, A. Metabolic Plasticity of Tumor Cell Mitochondria. Front. Oncol. 2018, 8, 333.
- Masgras, I.; Ciscato, F.; Brunati, A.M.; Tibaldi, E.; Indraccolo, S.; Curtarello, M.; Chiara, F.; Cannino, G.; Papaleo, E.; Lambrughi, M.; et al. Absence of Neurofibromin Induces an Oncogenic Metabolic Switch via Mitochondrial ERK-Mediated Phosphorylation of the Chaperone TRAP1. Cell Rep. 2017, 18, 659–672.
- Masgras, I.; Laquatra, C.; Cannino, G.; Serapian, S.A.; Colombo, G.; Rasola, A. The Molecular Chaperone TRAP1 in Cancer: From the Basics of Biology to Pharmacological Targeting. Semin. Cancer Biol. 2021, 76, 45–53.
- Porter, G.A.; Beutner, G. Cyclophilin D, Somehow a Master Regulator of Mitochondrial Function. Biomolecules 2018, 8, 176.
- Altieri, D.C.; Stein, G.S.; Lian, J.B.; Languino, L.R. TRAP-1, the Mitochondrial Hsp90. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2012, 1823, 767–773.
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 Chaperone Machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360.
- Obermann, W.M.J.; Sondermann, H.; Russo, A.A.; Pavletich, N.P.; Hartl, F.U. In Vivo Function of Hsp90 Is Dependent on ATP Binding and ATP Hydrolysis. J. Cell Biol. 1998, 143, 901–910.
- Panaretou, B. ATP Binding and Hydrolysis Are Essential to the Function of the Hsp90 Molecular Chaperone Invivo. EMBO J. 1998, 17, 4829–4836.
- Sahasrabudhe, P.; Rohrberg, J.; Biebl, M.M.; Rutz, D.A.; Buchner, J. The Plasticity of the Hsp90 Co-Chaperone System. Mol. Cell 2017, 67, 947–961.e5.
- Lavery, L.A.; Partridge, J.R.; Ramelot, T.A.; Elnatan, D.; Kennedy, M.A.; Agard, D.A. Structural Asymmetry in the Closed State of Mitochondrial Hsp90 (TRAP1) Supports a Two-Step ATP Hydrolysis Mechanism. Mol. Cell 2014, 53, 330–343.
- Partridge, J.R.; Lavery, L.A.; Elnatan, D.; Naber, N.; Cooke, R.; Agard, D.A. A Novel N-Terminal Extension in Mitochondrial TRAP1 Serves as a Thermal Regulator of Chaperone Activity. eLife 2014, 3, e03487.
- Ali, M.M.U.; Roe, S.M.; Vaughan, C.K.; Meyer, P.; Panaretou, B.; Piper, P.W.; Prodromou, C.; Pearl, L.H. Crystal Structure of an Hsp90–Nucleotide–P23/Sba1 Closed Chaperone Complex. Nature 2006, 440, 1013–1017.
- Mayer, M.P.; Le Breton, L. Hsp90: Breaking the Symmetry. Mol. Cell 2015, 58, 8–20.
- Mollapour, M.; Bourboulia, D.; Beebe, K.; Woodford, M.R.; Polier, S.; Hoang, A.; Chelluri, R.; Li, Y.; Guo, A.; Lee, M.-J.; et al. Asymmetric Hsp90 N Domain SUMOylation Recruits Aha1 and ATP-Competitive Inhibitors. Mol. Cell 2014, 53, 317–329.
- Retzlaff, M.; Hagn, F.; Mitschke, L.; Hessling, M.; Gugel, F.; Kessler, H.; Richter, K.; Buchner, J. Asymmetric Activation of the Hsp90 Dimer by Its Cochaperone Aha1. Mol. Cell 2010, 37, 344–354.
- Vaughan, C.K.; Gohlke, U.; Sobott, F.; Good, V.M.; Ali, M.M.U.; Prodromou, C.; Robinson, C.V.; Saibil, H.R.; Pearl, L.H. Structure of an Hsp90-Cdc37-Cdk4 Complex. Mol. Cell 2006, 23, 697–707.
- Elnatan, D.; Betegon, M.; Liu, Y.; Ramelot, T.; Kennedy, M.A.; Agard, D.A. Symmetry Broken and Rebroken during the ATP Hydrolysis Cycle of the Mitochondrial Hsp90 TRAP1. eLife 2017, 6, e25235.
- Joshi, A.; Dai, L.; Liu, Y.; Lee, J.; Ghahhari, N.M.; Segala, G.; Beebe, K.; Jenkins, L.M.; Lyons, G.C.; Bernasconi, L.; et al. The Mitochondrial HSP90 Paralog TRAP1 Forms an OXPHOS-Regulated Tetramer and Is Involved in Mitochondrial Metabolic Homeostasis. BMC Biol. 2020, 18, 10.
- Park, H.-K.; Hong, J.-H.; Oh, Y.T.; Kim, S.S.; Yin, J.; Lee, A.-J.; Chae, Y.C.; Kim, J.H.; Park, S.-H.; Park, C.-K.; et al. Interplay between TRAP1 and Sirtuin-3 Modulates Mitochondrial Respiration and Oxidative Stress to Maintain Stemness of Glioma Stem Cells. Cancer Res 2019, 79, 1369–1382.
- Lisanti, S.; Garlick, D.S.; Bryant, K.G.; Tavecchio, M.; Mills, G.B.; Lu, Y.; Kossenkov, A.V.; Showe, L.C.; Languino, L.R.; Altieri, D.C. Transgenic Expression of the Mitochondrial Chaperone TNFR-Associated Protein 1 (TRAP1) Accelerates Prostate Cancer Development. J. Biol. Chem. 2016, 291, 25247–25254.
- Ramkumar, B.; Dharaskar, S.P.; Mounika, G.; Paithankar, K.; Sreedhar, A.S. Mitochondrial Chaperone, TRAP1 as a Potential Pharmacological Target to Combat Cancer Metabolism. Mitochondrion 2020, 50, 42–50.
- Si, T.; Yang, G.; Qiu, X.; Luo, Y.; Liu, B.; Wang, B. Expression of Tumor Necrosis Factor Receptor-Associated. Int. J. Clin. Exp. Pathol. 2015, 8, 13090–13095.
- Zhang, B.; Wang, J.; Huang, Z.; Wei, P.; Liu, Y.; Hao, J.; Zhao, L.; Zhang, F.; Tu, Y.; Wei, T. Aberrantly Upregulated TRAP1 Is Required for Tumorigenesis of Breast Cancer. Oncotarget 2015, 6, 44495–44508.
- Xie, S. The Mitochondrial Chaperone TRAP1 as a Candidate Target of Oncotherapy. Front. Oncol. 2021, 10, 11.
- Serapian, S.A.; Sanchez-Martín, C.; Moroni, E.; Rasola, A.; Colombo, G. Targeting the Mitochondrial Chaperone TRAP1: Strategies and Therapeutic Perspectives. Trends Pharmacol. Sci. 2021, 42, 566–576.
- Miyata, Y.; Nakamoto, H.; Neckers, L. The Therapeutic Target Hsp90 and Cancer Hallmarks. Curr. Pharm. Des. 2013, 19, 347–365.
- Trepel, J.; Mollapour, M.; Giaccone, G.; Neckers, L. Targeting the Dynamic HSP90 Complex in Cancer. Nat. Rev. Cancer 2010, 10, 537–549.
- Zong, H.; Gozman, A.; Caldas-Lopes, E.; Taldone, T.; Sturgill, E.; Brennan, S.; Ochiana, S.O.; Gomes-DaGama, E.M.; Sen, S.; Rodina, A.; et al. A Hyperactive Signalosome in Acute Myeloid Leukemia Drives Addiction to a Tumor-Specific Hsp90 Species. Cell Rep. 2015, 13, 2159–2173.
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033.
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA Cycle Metabolites Control Physiology and Disease. Nat. Commun. 2020, 11, 102.
- Woodford, M.R.; Baker-Williams, A.J.; Sager, R.A.; Backe, S.J.; Blanden, A.R.; Hashmi, F.; Kancherla, P.; Gori, A.; Loiselle, D.R.; Castelli, M.; et al. The Tumor Suppressor Folliculin Inhibits Lactate Dehydrogenase A and Regulates the Warburg Effect. Nat. Struct. Mol. Biol. 2021, 28, 662–670.
- DeBerardinis, R.J.; Chandel, N.S. We Need to Talk about the Warburg Effect. Nat. Metab. 2020, 2, 127–129.
- Warburg, O. The Metabolism of Carcinoma Cells. J. Cancer Res. 1925, 9, 148–163.
- Sanchez-Martin, C.; Moroni, E.; Ferraro, M.; Laquatra, C.; Cannino, G.; Masgras, I.; Negro, A.; Quadrelli, P.; Rasola, A.; Colombo, G. Rational Design of Allosteric and Selective Inhibitors of the Molecular Chaperone TRAP1. Cell Rep. 2020, 31, 107531.
- Sanchez-Martin, C.; Menon, D.; Moroni, E.; Ferraro, M.; Masgras, I.; Elsey, J.; Arbiser, J.L.; Colombo, G.; Rasola, A. Honokiol Bis-Dichloroacetate Is a Selective Allosteric Inhibitor of the Mitochondrial Chaperone TRAP1. Antioxid. Redox Signal. 2021, 34, 505–516.
- Sciacovelli, M.; Guzzo, G.; Morello, V.; Frezza, C.; Zheng, L.; Nannini, N.; Calabrese, F.; Laudiero, G.; Esposito, F.; Landriscina, M.; et al. The Mitochondrial Chaperone TRAP1 Promotes Neoplastic Growth by Inhibiting Succinate Dehydrogenase. Cell Metab. 2013, 17, 988–999.
- Serapian, S.A.; Moroni, E.; Ferraro, M.; Colombo, G. Atomistic Simulations of the Mechanisms of the Poorly Catalytic Mitochondrial Chaperone Trap1: Insights into the Effects of Structural Asymmetry on Reactivity. ACS Catal. 2021, 11, 8605–8620.
- Xiang, F.; Ma, S.; Lv, Y.; Zhang, D.; Song, H.; Huang, Y. Tumor Necrosis Factor Receptor-Associated Protein 1 Regulates Hypoxia-Induced Apoptosis through a Mitochondria-Dependent Pathway Mediated by Cytochrome c Oxidase Subunit II. Burn. Trauma 2019, 7, s41038-019-0154-3s41038–s019.
- Xiang, F.; Ma, S.-Y.; Zhang, D.-X.; Zhang, Q.; Huang, Y.-S. Tumor Necrosis Factor Receptor-Associated Protein 1 Improves Hypoxia-Impaired Energy Production in Cardiomyocytes through Increasing Activity of Cytochrome c Oxidase Subunit II. Int. J. Biochem. Cell Biol. 2016, 79, 239–248.
- Bezawork-Geleta, A.; Rohlena, J.; Dong, L.; Pacak, K.; Neuzil, J. Mitochondrial Complex II: At the Crossroads. Trends Biochem. Sci. 2017, 42, 312–325.
- Liu, Y.; Elnatan, D.; Sun, M.; Myasnikov, A.G.; Agard, D.A. Cryo-EM Reveals the Dynamic Interplay between Mitochondrial Hsp90 and SdhB Folding Intermediates. bioRxiv 2020.
- Agarwal, E.; Altman, B.J.; Seo, J.H.; Ghosh, J.C.; Kossenkov, A.V.; Tang, H.-Y.; Krishn, S.R.; Languino, L.R.; Gabrilovich, D.I.; Speicher, D.W.; et al. Myc-Mediated Transcriptional Regulation of the Mitochondrial Chaperone TRAP1 Controls Primary and Metastatic Tumor Growth. J. Biol. Chem. 2019, 294, 10407–10414.
- Chae, Y.C.; Angelin, A.; Lisanti, S.; Kossenkov, A.V.; Speicher, K.D.; Wang, H.; Powers, J.F.; Tischler, A.S.; Pacak, K.; Fliedner, S.; et al. Landscape of the Mitochondrial Hsp90 Metabolome in Tumours. Nat. Commun. 2013, 4, 2139.
- Hu, S.; Ferraro, M.; Thomas, A.P.; Chung, J.M.; Yoon, N.G.; Seol, J.-H.; Kim, S.; Kim, H.; An, M.Y.; Ok, H.; et al. Dual Binding to Orthosteric and Allosteric Sites Enhances the Anticancer Activity of a TRAP1-Targeting Drug. J. Med. Chem. 2020, 63, 2930–2940.
- Masgras, I.; Sanchez-Martin, C.; Colombo, G.; Rasola, A. The Chaperone TRAP1 As a Modulator of the Mitochondrial Adaptations in Cancer Cells. Front. Oncol. 2017, 7, 58.
- Rizza, S.; Montagna, C.; Cardaci, S.; Maiani, E.; Di Giacomo, G.; Sanchez-Quiles, V.; Blagoev, B.; Rasola, A.; De Zio, D.; Stamler, J.S.; et al. S-Nitrosylation of the Mitochondrial Chaperone TRAP1 Sensitizes Hepatocellular Carcinoma Cells to Inhibitors of Succinate Dehydrogenase. Cancer Res. 2016, 76, 4170–4182.
- Fitzgerald, J.C.; Zimprich, A.; Carvajal Berrio, D.A.; Schindler, K.M.; Maurer, B.; Schulte, C.; Bus, C.; Hauser, A.-K.; Kübler, M.; Lewin, R.; et al. Metformin Reverses TRAP1 Mutation-Associated Alterations in Mitochondrial Function in Parkinson’s Disease. Brain 2017, 140, 2444–2459.
- Guzzo, G.; Sciacovelli, M.; Bernardi, P.; Rasola, A. Inhibition of Succinate Dehydrogenase by the Mitochondrial Chaperone TRAP1 Has Anti-Oxidant and Anti-Apoptotic Effects on Tumor Cells. Oncotarget 2014, 5, 11897–11908.
- Hsu, C.-C.; Tseng, L.-M.; Lee, H.-C. Role of Mitochondrial Dysfunction in Cancer Progression. Exp. Biol. Med. 2016, 241, 1281–1295.
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate Links TCA Cycle Dysfunction to Oncogenesis by Inhibiting HIF-α Prolyl Hydroxylase. Cancer Cell 2005, 7, 77–85.
- Zong, S.; Wu, M.; Gu, J.; Liu, T.; Guo, R.; Yang, M. Structure of the Intact 14-Subunit Human Cytochrome c Oxidase. Cell Res. 2018, 28, 1026–1034.
- Lisanti, S.; Tavecchio, M.; Chae, Y.C.; Liu, Q.; Brice, A.K.; Thakur, M.L.; Languino, L.R.; Altieri, D.C. Deletion of the Mitochondrial Chaperone TRAP-1 Uncovers Global Reprogramming of Metabolic Networks. Cell Rep. 2014, 8, 671–677.
- Sullivan, L.B.; Chandel, N.S. Mitochondrial Reactive Oxygen Species and Cancer. Cancer Metab. 2014, 2, 17.
- Clarke, B.E.; Kalmar, B.; Greensmith, L. Enhanced Expression of TRAP1 Protects Mitochondrial Function in Motor Neurons under Conditions of Oxidative Stress. Int. J. Mol. Sci. 2022, 23, 1789.
- Kang, B.H.; Plescia, J.; Dohi, T.; Rosa, J.; Doxsey, S.J.; Altieri, D.C. Regulation of Tumor Cell Mitochondrial Homeostasis by an Organelle-Specific Hsp90 Chaperone Network. Cell 2007, 131, 257–270.
- Leav, I.; Plescia, J.; Goel, H.L.; Li, J.; Jiang, Z.; Cohen, R.J.; Languino, L.R.; Altieri, D.C. Cytoprotective Mitochondrial Chaperone TRAP-1 As a Novel Molecular Target in Localized and Metastatic Prostate Cancer. Am. J. Pathol. 2010, 176, 393–401.
- Vartholomaiou, E.; Madon-Simon, M.; Hagmann, S.; Mühlebach, G.; Wurst, W.; Floss, T.; Picard, D. Cytosolic Hsp90α and Its Mitochondrial Isoform Trap1 Are Differentially Required in a Breast Cancer Model. Oncotarget 2017, 8, 17428–17442.
- Linehan, W.M.; Schmidt, L.S.; Crooks, D.R.; Wei, D.; Srinivasan, R.; Lang, M.; Ricketts, C.J. The Metabolic Basis of Kidney Cancer. Cancer Discov. 2019, 9, 1006–1021.
- Semenza, G.L. HIF-1: Upstream and Downstream of Cancer Metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56.
- Kowalik, M.A.; Guzzo, G.; Morandi, A.; Perra, A.; Menegon, S.; Masgras, I.; Trevisan, E.; Angioni, M.M.; Fornari, F.; Quagliata, L.; et al. Metabolic Reprogramming Identifies the Most Aggressive Lesions at Early Phases of Hepatic Carcinogenesis. Oncotarget 2016, 7, 32375–32393.
- Iacobazzi, V.; Infantino, V. Citrate—New Functions for an Old Metabolite. Biol. Chem. 2014, 395, 387–399.
- Liu, J.; Peng, Y.; Wei, W. Cell Cycle on the Crossroad of Tumorigenesis and Cancer Therapy. Trends Cell Biol. 2022, 32, 30–44.
- Amoroso, M.R.; Matassa, D.S.; Laudiero, G.; Egorova, A.V.; Polishchuk, R.S.; Maddalena, F.; Piscazzi, A.; Paladino, S.; Sarnataro, D.; Garbi, C.; et al. TRAP1 and the Proteasome Regulatory Particle TBP7/Rpt3 Interact in the Endoplasmic Reticulum and Control Cellular Ubiquitination of Specific Mitochondrial Proteins. Cell Death Differ. 2012, 19, 592–604.
- Sisinni, L.; Maddalena, F.; Condelli, V.; Pannone, G.; Simeon, V.; Li Bergolis, V.; Lopes, E.; Piscazzi, A.; Matassa, D.S.; Mazzoccoli, C.; et al. TRAP1 Controls Cell Cycle G2-M Transition through the Regulation of CDK1 and MAD2 Expression/Ubiquitination: TRAP1 Regulates Mitotic Entry through CDK1 Quality Control. J. Pathol. 2017, 243, 123–134.
- Liu, X.; Kim, C.N.; Yang, J.; Jemmerson, R.; Wang, X. Induction of Apoptotic Program in Cell-Free Extracts: Requirement for DATP and Cytochrome c. Cell 1996, 86, 147–157.
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and DATP-Dependent Formation of Apaf-1/Caspase-9 Complex Initiates an Apoptotic Protease Cascade. Cell 1997, 91, 479–489.
- Javadov, S.; Chapa-Dubocq, X.; Makarov, V. Different Approaches to Modeling Analysis of Mitochondrial Swelling. Mitochondrion 2018, 38, 58–70.
- Orrenius, S.; Gogvadze, V.; Zhivotovsky, B. Calcium and Mitochondria in the Regulation of Cell Death. Biochem. Biophys. Res. Commun. 2015, 460, 72–81.
- Bernardi, P.; Rasola, A.; Forte, M.; Lippe, G. The Mitochondrial Permeability Transition Pore: Channel Formation by F-ATP Synthase, Integration in Signal Transduction, and Role in Pathophysiology. Physiol. Rev. 2015, 95, 1111–1155.
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of Apoptosis Signalling Pathways by Reactive Oxygen Species. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2016, 1863, 2977–2992.
- Hua, G.; Zhang, Q.; Fan, Z. Heat Shock Protein 75 (TRAP1) Antagonizes Reactive Oxygen Species Generation and Protects Cells from Granzyme M-Mediated Apoptosis. J. Biol. Chem. 2007, 282, 20553–20560.
- Im, C.-N.; Lee, J.-S.; Zheng, Y.; Seo, J.-S. Iron Chelation Study in a Normal Human Hepatocyte Cell Line Suggests That Tumor Necrosis Factor Receptor-Associated Protein 1 (TRAP1) Regulates Production of Reactive Oxygen Species. J. Cell. Biochem. 2007, 100, 474–486.
- Costantino, E.; Maddalena, F.; Calise, S.; Piscazzi, A.; Tirino, V.; Fersini, A.; Ambrosi, A.; Neri, V.; Esposito, F.; Landriscina, M. TRAP1, a Novel Mitochondrial Chaperone Responsible for Multi-Drug Resistance and Protection from Apoptotis in Human Colorectal Carcinoma Cells. Cancer Lett. 2009, 279, 39–46.
- Landriscina, M.; Laudiero, G.; Maddalena, F.; Amoroso, M.R.; Piscazzi, A.; Cozzolino, F.; Monti, M.; Garbi, C.; Fersini, A.; Pucci, P.; et al. Mitochondrial Chaperone Trap1 and the Calcium Binding Protein Sorcin Interact and Protect Cells against Apoptosis Induced by Antiblastic Agents. Cancer Res. 2010, 70, 6577–6586.
- Suarez, J.; McDonough, P.M.; Scott, B.T.; Suarez-Ramirez, A.; Wang, H.; Fricovsky, E.S.; Dillmann, W.H. Sorcin Modulates Mitochondrial Ca2+ Handling and Reduces Apoptosis in Neonatal Rat Cardiac Myocytes. Am. J. Physiol. -Cell Physiol. 2013, 304, C248–C256.
- Elnatan, D.; Agard, D.A. Calcium Binding to a Remote Site Can Replace Magnesium as Cofactor for Mitochondrial Hsp90 (TRAP1) ATPase Activity. J. Biol. Chem. 2018, 293, 13717–13724.
- Zhang, L.; Su, N.; Luo, Y.; Chen, S.; Zhao, T. TRAP1 Inhibits MIC60 Ubiquitination to Mitigate the Injury of Cardiomyocytes and Protect Mitochondria in Extracellular Acidosis. Cell Death Discov. 2021, 7, 389.
- Friedman, J.R.; Mourier, A.; Yamada, J.; McCaffery, J.M.; Nunnari, J. MICOS Coordinates with Respiratory Complexes and Lipids to Establish Mitochondrial Inner Membrane Architecture. eLife 2015, 4, e07739.
- Rampelt, H.; Zerbes, R.M.; van der Laan, M.; Pfanner, N. Role of the Mitochondrial Contact Site and Cristae Organizing System in Membrane Architecture and Dynamics. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2017, 1864, 737–746.
- Baines, C.P.; Gutiérrez-Aguilar, M. The Mitochondrial Permeability Transition Pore: Is It Formed by the ATP Synthase, Adenine Nucleotide Translocators or Both? Biochim. Et Biophys. Acta (BBA)-Bioenerg. 2020, 1861, 148249.
- Carrer, A.; Tommasin, L.; Šileikytė, J.; Ciscato, F.; Filadi, R.; Urbani, A.; Forte, M.; Rasola, A.; Szabò, I.; Carraro, M.; et al. Defining the Molecular Mechanisms of the Mitochondrial Permeability Transition through Genetic Manipulation of F-ATP Synthase. Nat. Commun. 2021, 12, 4835.
- Giorgio, V.; von Stockum, S.; Antoniel, M.; Fabbro, A.; Fogolari, F.; Forte, M.; Glick, G.D.; Petronilli, V.; Zoratti, M.; Szabó, I.; et al. Dimers of Mitochondrial ATP Synthase Form the Permeability Transition Pore. Proc. Natl. Acad. Sci. USA 2013, 110, 5887–5892.
- Kang, B.H.; Tavecchio, M.; Goel, H.L.; Hsieh, C.-C.; Garlick, D.S.; Raskett, C.M.; Lian, J.B.; Stein, G.S.; Languino, L.R.; Altieri, D.C. Targeted Inhibition of Mitochondrial Hsp90 Suppresses Localised and Metastatic Prostate Cancer Growth in a Genetic Mouse Model of Disease. Br. J. Cancer 2011, 104, 629–634.
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of Cyclophilin D Reveals a Critical Role for Mitochondrial Permeability Transition in Cell Death. Nature 2005, 434, 658–662.
- Matassa, D.S.; Amoroso, M.R.; Maddalena, F.; Landriscina, M. New Insights into TRAP1 Pathway. Am. J. Cancer Res. 2012, 2, 235–248.
- Ghosh, J.C.; Siegelin, M.D.; Dohi, T.; Altieri, D.C. Heat Shock Protein 60 Regulation of the Mitochondrial Permeability Transition Pore in Tumor Cells. Cancer Res. 2010, 70, 8988–8993.
- Lebedev, I.; Nemajerova, A.; Foda, Z.H.; Kornaj, M.; Tong, M.; Moll, U.M.; Seeliger, M.A. A Novel In Vitro CypD-Mediated P53 Aggregation Assay Suggests a Model for Mitochondrial Permeability Transition by Chaperone Systems. J. Mol. Biol. 2016, 428, 4154–4167.
- Sinha, D.; D’Silva, P. Chaperoning Mitochondrial Permeability Transition: Regulation of Transition Pore Complex by a J-Protein, DnaJC15. Cell Death Dis. 2014, 5, e1101.
- Niemi, N.M.; Pagliarini, D.J. The Extensive and Functionally Uncharacterized Mitochondrial Phosphoproteome. J. Biol. Chem. 2021, 297, 100880.
- Faienza, F.; Lambrughi, M.; Rizza, S.; Pecorari, C.; Giglio, P.; Salamanca Viloria, J.; Allega, M.F.; Chiappetta, G.; Vinh, J.; Pacello, F.; et al. S-Nitrosylation Affects TRAP1 Structure and ATPase Activity and Modulates Cell Response to Apoptotic Stimuli. Biochem. Pharmacol. 2020, 176, 113869.
- Rasola, A.; Neckers, L.; Picard, D. Mitochondrial Oxidative Phosphorylation TRAP(1)Ped in Tumor Cells. Trends Cell Biol. 2014, 24, 455–463.
- Cloutier, P.; Coulombe, B. Regulation of Molecular Chaperones through Post-Translational Modifications: Decrypting the Chaperone Code. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2013, 1829, 443–454.
- Zuehlke, A.D.; Reidy, M.; Lin, C.; LaPointe, P.; Alsomairy, S.; Lee, D.J.; Rivera-Marquez, G.M.; Beebe, K.; Prince, T.; Lee, S.; et al. An Hsp90 Co-Chaperone Protein in Yeast Is Functionally Replaced by Site-Specific Posttranslational Modification in Humans. Nat. Commun. 2017, 8, 15328.

