Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Laura Cortesi | -- | 2785 | 2022-06-07 09:53:12 | | | |
| 2 | Vivi Li | Meta information modification | 2785 | 2022-06-07 11:57:55 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Cortesi, L.; Piombino, C. Molecular Mechanisms of Resistance to PARP Inhibitors. Encyclopedia. Available online: https://encyclopedia.pub/entry/23765 (accessed on 27 June 2026).
Cortesi L, Piombino C. Molecular Mechanisms of Resistance to PARP Inhibitors. Encyclopedia. Available at: https://encyclopedia.pub/entry/23765. Accessed June 27, 2026.
Cortesi, Laura, Claudia Piombino. "Molecular Mechanisms of Resistance to PARP Inhibitors" Encyclopedia, https://encyclopedia.pub/entry/23765 (accessed June 27, 2026).
Cortesi, L., & Piombino, C. (2022, June 07). Molecular Mechanisms of Resistance to PARP Inhibitors. In Encyclopedia. https://encyclopedia.pub/entry/23765
Cortesi, Laura and Claudia Piombino. "Molecular Mechanisms of Resistance to PARP Inhibitors." Encyclopedia. Web. 07 June, 2022.
Copy Citation
PARP1 enzyme plays an important role in DNA damage recognition and signalling. PARP inhibitors are approved in breast, ovarian, pancreatic, and prostate cancers harbouring a pathogenic variant in BRCA1 or BRCA2, where PARP1 inhibition results mainly in synthetic lethality in cells with impaired homologous recombination. However, the increasingly wide use of PARP inhibitors in clinical practice has highlighted the problem of resistance to therapy. Several different mechanisms of resistance have been proposed, although only the acquisition of secondary mutations in BRCA1/2 has been clinically proved.
PARP inhibitors
BRCA1
BRCA2
homologous recombination
non-homologous end joining
fork stabilization
1. Introduction
Poly-(ADP-ribose) polymerase (PARP) enzyme PARP1 plays an important role in DNA damage recognition and signalling, as it binds single-stranded DNA breaks (SSBs) and then organizes their repair by synthesising PAR chains on target proteins (the so-called PARylation) [1]. In detail PARP1, once bound to SSBs via N-terminal zinc-finger DNA-binding domain, catalyses the polymerization of ADP-ribose moieties from NAD+ on target proteins, mainly PARP1 itself and histones. This process leads to chromatin relaxation and recruitment of other DNA repair enzymes such as XRCC1 [2][3][4][5]. The scaffolding protein XRCC1 stimulates the DNA kinase and DNA phosphatase activities of polynucleotide kinase at SSBs, accelerating the base excision repair reaction [6]. It is also reported that PARP1 promotes recruiting of MRE11, ATM and BRCA1, which are involved in double-stranded DNA break (DSB) repair by homologous recombination (HR) [7][8][9]. While PARP1 DNA binding is independent of its catalytic activity, dissociation of PARP1 from DNA requires PARylation presumably through a steric mechanism due to highly negatively charged PAR chains [10].
PARP inhibitors (PARPi) act mainly in a double way. The first proposed mechanism is the inhibition of the catalytic activity of PARP1, which results in synthetic lethality in cells with impaired HR [11][12][13]. In fact, inhibition of PARP1 promotes SSBs, which, if unrepaired, consequently lead to DSBs by collapse of the stalled replication fork during DNA replication [14]. In eukaryotic cells, DSBs are mainly resolved by the error-free mechanism of HR, which uses the intact sister chromatid as a template. HR deficiency induces activation of the more error-prone template-independent non-homologous end-joining (NHEJ) pathway, therefore, together with PARPi causing genomic instability followed by cell death [15][16]. Subsequent studies have revealed that most PARPi cause cytotoxicity by trapping PARP1 at sites of DNA damage [17][18][19]. According to the hypothesis proposed by Murai et al. [18], PARPi binding to the catalytic domain of PARP1 allosterically modifies interactions between DNA and the N-terminal DNA-binding domain of the protein, to the point that PARP1 becomes trapped on DNA. More recently, a third mechanism of PARPi sensitivity has been identified [20][21]. In cells with HR deficiency, aside from the NHEJ pathway, DSBs can be repaired by the microhomology-mediated end joining (MMEJ or Alt-EJ) pathway. Similarly to NHEJ, MMEJ is intrinsically error-prone, as the use of regions of microhomology inside DNA leads to deletions of nucleotides from the strand being repaired and to chromosomal translocations. In this pathway, the efficient recruitment of the DNA polymerase POLQ to the DSB requires PARP1. A PARPi will, therefore, block the MMEJ pathway and cause HR-deficient cell death.
PARP inhibitors are actually approved in breast, ovarian, pancreatic and prostate cancers harbouring a pathogenic or likely pathogenic variant in BRCA1 or BRCA2 (BRCA1/2) [22][23][24]. BRCA1/2 mutation prevalence varied widely from 1.8% in sporadic breast cancer to 36.9% in estrogen receptor/progesterone receptor low HER2 negative breast cancer [25]. Germline mutations in BRCA1/2 have been identified in 13–15% of women diagnosed with ovarian cancer, and somatic mutations are found in an additional 7% [26][27][28]. Germline BRCA1/2 mutations can be found in up to 8% of patients with sporadic pancreatic cancer [29]. In a sample of 692 patients with metastatic prostate cancer, unselected for family history or age at diagnosis, 5.3% carried a BRCA2 mutation, and 0.9% carried a BRCA1 mutation [30]. The increasingly wide use of PARPi in clinical practice is highlighting the problem of resistance to therapy. Considering the complex interaction between PARP1, HR and other DNA damage repair pathways in the setting of BRCA1/2 mutated cancers, several different mechanisms of resistance have been proposed, although some of them have been only described preclinically. The aim of this entry is to outline the key molecular findings that could explain the mechanisms of primary or secondary resistance to PARPi (summarised in Table 1).
Table 1. Proposed mechanisms of PARPi resistance.
| Resistance Mechanism | Evidence | References |
|---|---|---|
| Primary resistance | ||
| PI3K/AKT pathway activation | Cell lines | Yi et al. [31] |
| Wild-type PTEN | Cell lines | Dedes et al. [32] |
| Loss of NHEJ | Cell lines | Balmus et al. [33], Patel et al. [34], Mc Cormick et al. [35] |
| ALC1 overexpression | Cell lines | Juhász et al. [36] |
| Secondary resistance | ||
| Upregulation of ABC transporters | Mouse models, cell lines |
Jaspers et al. [37], Vaidyanathan et al. [38] |
| Decreased PARP1 trapping | Mouse models, cell lines |
Pettitt et al. [39], Gogola et al. [40] |
| Restoration of HR | ||
| -BRCA1/2 reversion mutations | Tumour DNA and ctDNA from cancer patients | Tobalina et al. [41] |
| -Hypomorphic BRCA1 allele | Cell lines, mouse models, PDXs | Drost et al. [42], Wang et al. [43], Cruz et al. [44], Wang et al. [45], Castroviejo-Bermejo et al. [46] |
| -Loss of BRCA1 promoter methylation | Cell lines, PDXs | Ter Brugge et al. [47], Veeck et al. [48], Wang et al. [49] |
| -Loss of end resection regulation (53BP1, RIF1, REV7, Sheldin complex or DYNLL1 depletion) | Cell lines | Belotserkovskaya et al. [50], Xu et al. [51], Noordermeer et al. [52], Gupta et al. [53], He et al. [54] |
| -RAD51 overexpression | Ovarian cancer samples, cell lines | Kondrashova et al. [55], Liu et al. [56], Marzio et al. [57] |
| Stabilization of stalled fork (FANCD2 overexpression, RADX depletion, SMARCAL1 inactivation,) | Cell lines | Michl et al. [58], Chaudhuri et al. [59], Taglialatela et al. [60], Dungrawala et al. [61] |
NHEJ: non-homologous end-joining. PDXs: patient-derived xenografts.
2. Primary Resistance
2.1. PTEN Deficiency and PI3K/AKT Pathway Activation
Phosphatase and tensin homolog (PTEN) is a dual protein/lipid phosphatase that acts as a tumour suppressor gene inhibiting the PI3K/AKT pathway. In detail, PTEN converts phosphatidylinositol 3,4,5-triphosphate (PIP3) into phosphatidylinositol 4,5-bisphosphate (PIP2), antagonising PI3K action and preventing AKT activation and consequent cell growth and cell proliferation [62][63]. PTEN is often inactivated in different cancers [64]. Although AKT activation promotes BRCA1 expression through phosphorylation [65], it has been shown that BRCA1 can downregulate AKT activation in different ways: by acting on upstream kinases of AKT [31][66], by ubiquitin-mediated proteasomal degradation or by activating a protein serine/threonine phosphatase PP2A in breast cancer cells (Figure 1) [67]. Therefore, negative mutations and/or reduced expression of BRCA1 may activate the PI3K/AKT pathway [67]. Significantly, PTEN loss is highly associated with BRCA1-defective breast cancers, probably due to genomic instability resulting from deficient DSB repair [68], and the resulting PI3K/AKT activation stimulates the growth of those cancers [69].
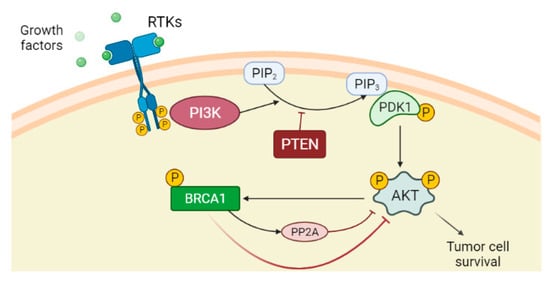
Figure 1. PI3K/AKT pathway is an intracellular signal transduction pathway that promotes cell growth and proliferation in response to extracellular signals. The binding of the ligands such as growth factors to the receptor tyrosine kinases (RTKs) induces dimerization of two RTK monomers, which consequently leads to activation of the intracellular tyrosine kinase domain and auto-phosphorylation by each monomer. The phosphatidylinositol 3-kinase (PI3K), once activated through direct stimulation of the regulatory subunit bound to the activated receptor, converts phosphatidylinositol 4,5-bisphosphate (PIP2) into phosphatidylinositol 3,4,5-triphosphate (PIP3). PIP3 binds the 3-phosphoinositide-dependent protein kinase-1 (PDK1) at the plasma membrane. PDK1 in turn phosphorylates and activates AKT protein. Once activated, AKT via phosphorylation regulates activation or suppression of several proteins involved in cell growth and proliferation. Phosphatase and tensin homolog (PTEN) is the main downregulation protein that can convert PIP3 into PIP2. Although AKT activation promotes BRCA1 expression through phosphorylation, BRCA1 can downregulate AKT activation by different mechanisms, among which are the activation of protein phosphatase 2A (PP2A), which dephosphorylates AKT.
Apart from its role in regulating the PI3K/AKT pathway, PTEN loss of function causes genetic instability, as PTEN-null cells lack competent HR DNA repair. In detail, PTEN has been suggested to contribute to RAD51 expression [70][71][72]. Dedes et al. [32] demonstrated that endometrioid endometrial cancer cell lines lacking PTEN function are unable to elicit competent HR DNA repair and are relatively sensitive to PARPi, and, as a consequence, PTEN silencing significantly increases the sensitivity to PARPi reducing RAD51 foci formation. Re-expression of PTEN in PTEN-mutant endometrioid endometrial cancer cells leads to relative resistance to PARPi [32].
Together, these observations corroborate the hypothesis that PTEN loss, which is highly associated with BRCA1-defective breast cancer, contributes to PARPi sensitivity. On the other hand, wild-type PTEN tumours could demonstrate relative resistance to PARPi. Considering that the PI3K/AKT pathway is constitutively active in BRCA1-defective human cancer cells [73], the combination of PTEN-related PI3K/AKT pathway inhibitors such as perifosine with DNA topoisomerase I (TOP1) or PARPi results in enhanced cancer cell killing in these tumours [74][75], suggesting a new possibly targetable pathway in case of PARPi resistance. A phase I trial of the PARPi Olaparib with the AKT inhibitor capivasertib, involving patients with advanced solid tumours carrying a germline BRCA1/2 mutation or somatic DNA damage response or PI3K/AKT pathway alterations, demonstrated the safety, tolerability and pharmacokinetic-pharmacodynamic activity of this combination [76].
2.2. ATM Roles and Loss of NHEJ Pathway
The choice between NHEJ and HR to repair DSBs is determined by several mechanisms, including activation of HR by cyclin-dependent kinase (CDK) activity (while NHEJ operates throughout interphase, HR is restricted to the S and G2 phase of the cell cycle, when a homologous sister chromatid is available as template) [16], or direct competition between HR and NHEJ stimulating factors at DSB sites [77]. During G2/S, HR is activated by binding of the MRE11–RAD50–NBS1 (MRN) complex to DSB ends. The MRN complex initiates DNA end resection, leading to the formation of single-strand DNA (ssDNA) at the extremity of the DSB. The ssDNA is protected from degradation by the loading of replication protein A (RPA) [78]. CtIP phosphorylated by CDK binds MRN complex to facilitate end resection [79]. The MRN complex recruits and activates the protein kinase ATM [80], while the sensor protein RPA finally drives ATR activation [81]. ATM and ATR phosphorylate several proteins involved in the HR [82]. The tumour suppressor complex BRCA1-BARD1 phosphorylated by ATM facilitates DNA end resection and interacts with PALB2, which in turn promotes the recruitment of BRCA2 [54]. PALB2 and BRCA2 remove RPA and facilitate the assembly of the RAD51 recombinase nucleoprotein filament. RAD51 nucleoprotein filament mediates the invasion of ssDNA into the intact sister chromatid, searching for a homologous template for DNA synthesis and repair [83][84][85]. During G1/2, 5′-3′ end resection is suppressed, and HR is inhibited due to the lack of sister chromatid [86]. ATM phosphorylates 53BP1 in multiple residues [87]. Once phosphorylated, 53BP1 binds and recruits RIF1 and PTIP that, together with the downstream effectors REV7 and Sheldin, inhibit 5′-3′ end resection and promote NHEJ [88]. Classical NHEJ starts when Ku70/80 heterodimer binds DSBs and recruits the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) to form the DNA–PK complex. The latter engages XRCC4 and DNA ligase IV (LIG4) to align and ligates DNA ends regardless of sequence homology [89]. BRCA1 antagonizes 53BP1 by limiting its interaction with the chromatin in the S phase and stopping the translocation of RIF1 to DSBs in the G2/S phase, promoting HR [90][91]. Additionally, the different recruitment kinetics of the MRN and Ku complexes, which activate HR and NHEJ repair, respectively [92], as well as MRN/CtIP-dependent removal of Ku complex from DSBs [93], influence the choice between the two pathways (Figure 2).
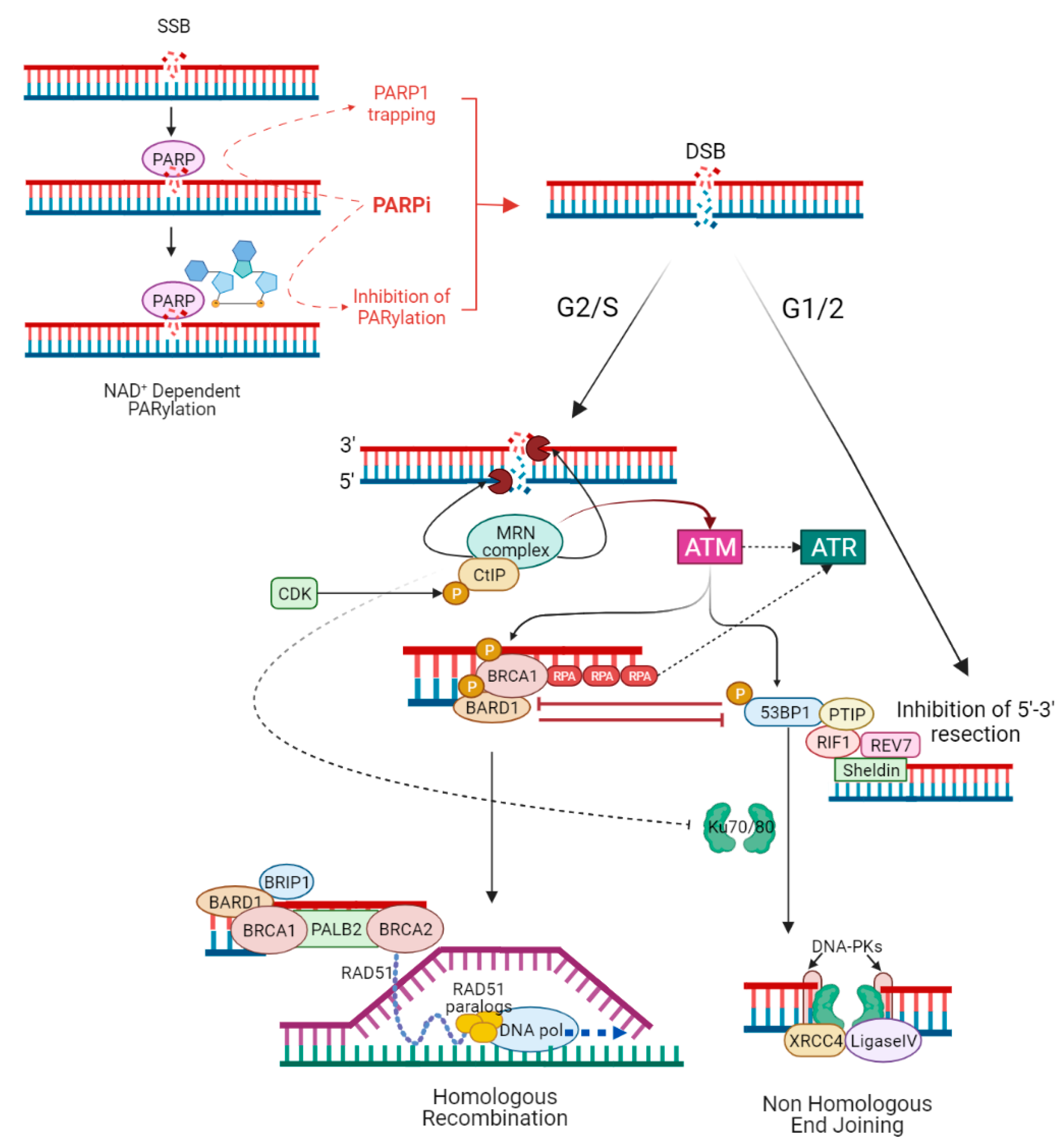
Figure 2. PARP inhibitors and the interactions between homologous recombination and non-homologous end joining. PARPi act mainly in a double way: inhibiting the catalytic activity of PARP1 (the so-called PARylation) and trapping PARP1 at sites of single-stranded DNA breaks (SSBs). In both cases, unrepaired SSBs lead to double-stranded DNA breaks (DSBs), which can be resolved mainly by HR or NHEJ. The choice between NHEJ and HR to repair DSBs is determined by several mechanisms, including activation of HR by cyclin-dependent kinase (CDK) activity (HR is restricted to G2/S phase when a homologous sister chromatid is available as template), or direct competition between HR and NHEJ stimulating factors at DSB sites. During G2/S, HR is activated by the binding of the MRN complex to DSB ends; MRN complex initiates DNA 5′-3′ end resection, leading to the formation of single-strand DNA (ssDNA) at the extremity of the DSB. CDK phosphorylated CtIP binds MRN complex to facilitate end resection. The ssDNA is protected from degradation by the loading of replication protein A (RPA). The MRN complex recruits and activates the protein kinase ATM, while RPA finally drives ATR activation. ATM phosphorylated BRCA1–BARD1 complex interacts with the bridging protein PALB2, which in turn promotes the recruitment of BRCA2. PALB2 and BRCA2 remove RPA and facilitate the assembly of the RAD51 nucleoprotein filament. RAD51 nucleoprotein filament mediates the invasion of ssDNA into the intact sister chromatid, searching for a homologous template for DNA synthesis and faithful repair of DNA. During G1/2, 5′-3′ end resection is suppressed and HR is inhibited due to lack of a sister chromatid. ATM phosphorylated 53BP1 binds and recruits RIF1 and PTIP that, together with the downstream effectors REV7 and Sheldin, inhibit 5′-3′ end resection and promote NHEJ. Ku70/80 heterodimer binds DSBs and recruits the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) to form the DNA–PK complex. The latter engages XRCC4, XLF, and DNA ligase IV (LIG4) to align and ligates DNA ends regardless of sequence homology. BRCA1 antagonizes 53BP1 by stopping the translocation of RIF1 to DSBs in the G2/S phase, promoting HR; also, MRN/CtIP-dependent removal of Ku complex from DSBs favours HR.
2.3. ALC1 Overexpression
Amplified in liver cancer 1 (ALC1) is a PAR-dependent chromatin remodeler that directly binds PAR chains, promoting PARP1 activation [94][95]. In a genome-wide CRISPR knockout screen with Olaparib, Juhász and colleagues [36] identified ALC1 as a key modulator of sensitivity to the PARPi Olaparib. ALC1 deficiency stimulates trapping of inhibited PARP1, which then impairs the binding of both the HR and NHEJ repair factors to DNA lesions. Moreover, they established that ALC1 overexpression, which is a common trait of many solid cancers, often associated with poor prognosis [96], reduces the sensitivity of BRCA1/2-deficient cells to PARPi. Analysis of the ALC1 expression levels before the use of PARPi could predict a mechanism of primary resistance to the therapy.
3. Secondary Resistance
Secondary or acquired resistance can be defined as the onset of the lack of response to treatment despite being successfully used before. Under therapeutic selective pressure, resistance to cytotoxic or target agents can emerge due to the expansion of pre-existing subclonal populations or from the evolution of drug-tolerant cells [97]. Several mechanisms of acquired resistance to PARPi have been suggested, although only reversion mutations in BRCA1/2 have been proved in the clinical setting.
3.1. Upregulation of ABC Transporters
Increased expression of the membrane-bound ATP-dependent efflux pump P-glicoprotein, encoded by ABCB1 (MDR1), is one of the most well-characterised mechanisms of multidrug resistance [98], in particular for doxorubicin, paclitaxel and related taxane drugs [99][100][101]. A chromosomal amplification event at 7q11.2-21 has been correlated to increased ABCB1 copy number and consequent P-glycoprotein expression in paclitaxel-resistant cancer cells [102].
Using a mouse model of BRCA2-deficient sarcomatoid breast cancer, Jaspers et al. [37] found that multidrug resistance was strongly associated with high expression of the ABCB1, known to be efflux transporter of Olaparib [103], docetaxel [104], and doxorubicin [105]. In novel A2780-derived ovarian cancer cell lines, paclitaxel-resistant cells were cross-resistant to the PARPi Olaparib and Rucaparib but not to Veliparib. This resistance correlated with increased ABCB1 expression and was reversible following treatment with ABCB1 inhibitors [38]. These findings, although cell-line based, could help in PARPi choice in paclitaxel-resistant patients, especially in second-line or maintenance therapy.
3.2. Decreased PARP1 Trapping
Through a genome-wide and high-density CRISPR-Cas9 mutagenesis screen to identify mouse embryonic stem cell mutants resistant to the PARPi Talazoparib, Pettitt and colleagues [39] identified point mutations in the N-terminal zinc-finger domain of PARP1 that, abolishing DNA binding, cause PARP1 resistance and alter PARP1 trapping. In support of this finding, they also observed a PARP1 mutation that abolished trapping in a patient with de novo resistance to Olaparib. In this experimental model, also mutations in amino-acid residues involved in hydrogen-bonding interactions that bridge the DNA-binding domain and the catalytic domain can cause PARPi resistance, likely by impairing PARP1 trapping; this reinforces the observations that inter-domain interactions are critical for PARP1 binding and activation [2][3][4].
PAR glycohydrolase (PARG) is the main enzyme responsible for degrading PAR chains, counteracting PARylation catalysed by PARP1 [106]. By combining genetic screens with multi-omics analysis of matched PARPi sensitive and resistant BRCA2-mutated mouse mammary tumours, Gogola et al. [40] identified loss of PARG as a major resistance mechanism. In a panel of 34 PARPi resistant tumours, they observed decreased expression of Parg in 17 tumours and acquired copy-number loss of the Parg locus in 22 tumours. Moreover, they demonstrated that PARG depletion does not increase PARP1 trapping per se but prevents excessive PARP1 binding to chromatin, thus reducing PARPi-dependent accumulation of toxic PARP1–DNA complexes. As a confirmation of this, PARG depletion also resulted in resistance to Talazoparib, a highly potent PARP1–DNA trapping agent. Finally, they found PARG-negative cell clones in a subset of human serous ovarian and triple-negative breast cancers (TNBCs), suggesting that this mechanism can contribute to PARPi resistance in vivo.
References
- Mortusewicz, O.; Amé, J.C.; Schreiber, V.; Leonhardt, H. Feedback-regulated poly(ADP-ribosyl)ation by PARP-1 is required for rapid response to DNA damage in living cells. Nucleic Acids Res. 2007, 35, 7665–7675.
- Dawicki-McKenna, J.M.; Langelier, M.F.; DeNizio, J.E.; Riccio, A.A.; Cao, C.D.; Karch, K.R.; McCauley, M.; Steffen, J.D.; Black, B.E.; Pascal, J.M. PARP-1 Activation Requires Local Unfolding of an Autoinhibitory Domain. Mol. Cell 2015, 60, 755–768.
- Eustermann, S.; Wu, W.F.; Langelier, M.F.; Yang, J.C.; Easton, L.E.; Riccio, A.A.; Pascal, J.M.; Neuhaus, D. Structural Basis of Detection and Signaling of DNA Single-Strand Breaks by Human PARP-1. Mol. Cell 2015, 60, 742–754.
- Langelier, M.F.; Planck, J.L.; Roy, S.; Pascal, J.M. Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science 2012, 336, 728–732.
- El Khamisy, S.F.; Masutani, M.; Suzuki, H.; Caldecott, K.W. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003, 31, 5526–5533.
- Whitehouse, C.J.; Taylor, R.M.; Thistlethwaite, A.; Zhang, H.; Karimi-Busheri, F.; Lasko, D.D.; Weinfeld, M.; Caldecott, K.W. XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell 2001, 104, 107–117.
- Haince, J.F.; McDonald, D.; Rodrigue, A.; Dery, U.; Masson, J.Y.; Hendzel, M.J.; Poirier, G.G. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 2008, 283, 1197–1208.
- Haince, J.F.; Kozlov, S.; Dawson, V.L.; Dawson, T.M.; Hendzel, M.J.; Lavin, M.F.; Poirier, G.G. Ataxia telangiectasia mutated (ATM) signaling network is modulated by a novel poly(ADP-ribose)-dependent pathway in the early response to DNA-damaging agents. J. Biol. Chem. 2007, 282, 16441–16453.
- Li, M.; Yu, X. Function of BRCA1 in the DNA damage response is mediated by ADP-ribosylation. Cancer Cell 2013, 23, 693–704.
- Zahradka, P.; Ebisuzaki, K. A shuttle mechanism for DNA-protein interactions. The regulation of poly(ADP-ribose) polymerase. Eur. J. Biochem. 1982, 127, 579–585.
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921.
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917.
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158.
- Liao, H.; Ji, F.; Helleday, T.; Ying, S. Mechanisms for stalled replication fork stabilization: New targets for synthetic lethality strategies in cancer treatments. EMBO Rep. 2018, 19, e46263.
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294.
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 2012, 47, 497–510.
- Murai, J.; Huang, S.Y.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP trapping by BMN 673 and comparison with laparib and rucaparib. Mol. Cancer Ther. 2014, 13, 433–443.
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012, 72, 5588–5599.
- Pettitt, S.J.; Rehman, F.L.; Bajrami, I.; Brough, R.; Wallberg, F.; Kozarewa, I.; Fenwick, K.; Assiotis, I.; Chen, L.; Campbell, J.; et al. A genetic screen using the PiggyBac transposon in haploid cells identifies Parp1 as a mediator of laparib toxicity. PLoS ONE 2013, 8, e61520.
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.; O’Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature 2015, 518, 258–262.
- Mateos-Gomez, P.A.; Gong, F.; Nair, N.; Miller, K.M.; Lazzerini-Denchi, E.; Sfeir, A. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature 2015, 518, 254–257.
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327.
- De Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102.
- Mehta, P.; Bothra, S.J. PARP inhibitors in hereditary breast and ovarian cancer and other cancers: A review. Adv. Genet. 2021, 108, 35–80.
- Armstrong, N.; Ryder, S.; Forbes, C.; Ross, J.; Quek, R.G. A systematic review of the international prevalence of BRCA mutation in breast cancer. Clin. Epidemiol. 2019, 11, 543–561.
- Alsop, K.; Fereday, S.; Meldrum, C.; DeFazio, A.; Emmanuel, C.; George, J.; Dobrovic, A.; Birrer, M.J.; Webb, P.M.; Stewart, C.; et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: A report from the Australian Ovarian Cancer Study Group. J. Clin. Oncol. 2012, 30, 2654–2663.
- Pennington, K.P.; Walsh, T.; Harrell, M.I.; Lee, M.K.; Pennil, C.C.; Rendi, M.H.; Thornton, A.; Norquist, B.M.; Casadei, S.; Nord, A.S.; et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin. Cancer Res. 2014, 20, 764–775.
- Zhang, S.; Royer, R.; Li, S.; McLaughlin, J.R.; Rosen, B.; Risch, H.A.; Fan, I.; Bradley, L.; Shaw, P.A.; Narod, S.A. Frequencies of BRCA1 and BRCA2 mutations among 1342 unselected patients with invasive ovarian cancer. Gynecol. Oncol. 2011, 121, 353–357.
- Rosen, M.N.; Goodwin, R.A.; Vickers, M.M. BRCA mutated pancreatic cancer: A change is coming. World J. Gastroenterol. 2021, 27, 1943–1958.
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453.
- Yi, Y.W.; Kang, H.J.; Kim, H.J.; Hwang, J.S.; Wang, A.; Bae, I. Inhibition of constitutively activated phosphoinositide 3-kinase/AKT pathway enhances anti- tumor activity of chemotherapeutic agents in breast cancer susceptibility gene 1-defective breast cancer cells. Mol. Carcinog. 2013, 52, 667–675.
- Dedes, K.J.; Wetterskog, D.; Mendes-Pereira, A.M.; Natrajan, R.; Lambros, M.B.; Geyer, F.C.; Vatcheva, R.; Savage, K.; Mackay, A.; Lord, C.J.; et al. PTEN Deficiency in Endometrioid Endometrial Adenocarcinomas Predicts Sensitivity to PARP Inhibitors. Sci. Transl. Med. 2010, 2, 53ra75.
- Balmus, G.; Pilger, D.; Coates, J.; Demir, M.; Sczaniecka-Clift, M.; Barros, A.C.; Woods, M.; Fu, B.; Yang, F.; Chen, E.; et al. ATM orchestrates the DNA-damage response to counter toxic non-homologous end-joining at broken replication forks. Nat. Commun. 2019, 10, 87.
- Patel, A.G.; Sarkaria, J.N.; Kaufmann, S.H. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc. Natl. Acad. Sci. USA 2011, 108, 3406–3411.
- McCormick, A.; Donoghue, P.; Dixon, M.; O’Sullivan, R.; O’Donnell, R.L.; Murray, J.; Kaufmann, A.; Curtin, N.J.; Edmondson, R.J. Ovarian Cancers Harbor Defects in Nonhomologous End Joining Resulting in Resistance to Rucaparib. Clin. Cancer Res. 2017, 23, 2050–2060.
- Juhász, S.; Smith, R.; Schauer, T.; Spekhardt, D.; Mamar, H.; Zentout, S.; Chapuis, C.; Huet, S.; Timinszky, G. The chromatin remodeler ALC1 underlies resistance to PARP inhibitor treatment. Sci. Adv. 2020, 6, eabb8626.
- Jaspers, J.E.; Sol, W.; Kersbergen, A.; Schlicker, A.; Guyader, C.; Xu, G.; Wessels, L.; Borst, P.; Jonkers, J.; Rottenberg, S. BRCA2-deficient sarcomatoid mammary tumors exhibit multidrug resistance. Cancer Res. 2015, 75, 732–741.
- Vaidyanathan, A.; Sawers, L.; Gannon, A.L.; Chakravarty, P.; Scott, A.L.; Bray, S.E.; Ferguson, M.J.; Smith, G. ABCB1 (MDR1) induction defines a common resistance mechanism in paclitaxel- and olaparib-resistant ovarian cancer cells. Br. J. Cancer 2016, 115, 431–441.
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Dréan, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun. 2018, 9, 1849.
- Gogola, E.; Duarte, A.A.; de Ruiter, J.R.; Wiegant, W.W.; Schmid, J.A.; de Bruijn, R.; James, D.I.; Guerrero Llobet, S.; Vis, D.J.; Annunziato, S.; et al. Selective Loss of PARG Restores PARylation and Counteracts PARP Inhibitor-Mediated Synthetic Lethality. Cancer Cell 2018, 33, 1078–1093.e12.
- Tobalina, L.; Armenia, J.; Irving, E.; O’Connor, M.J.; Forment, J.V. A meta-analysis of reversion mutations in BRCA genes identifies signatures of DNA end-joining repair mechanisms driving therapy resistance. Ann. Oncol. 2021, 32, 103–112.
- Drost, R.; Dhillon, K.K.; van der Gulden, H.; van der Heijden, I.; Brandsma, I.; Cruz, C.; Chondronasiou, D.; Castroviejo-Bermejo, M.; Boon, U.; Schut, E.; et al. BRCA1185delAG tumors may acquire therapy resistance through expression of RING-less BRCA1. J. Clin. Investig. 2016, 126, 2903–2918.
- Wang, Y.; Bernhardy, A.J.; Cruz, C.; Krais, J.J.; Nacson, J.; Nicolas, E.; Peri, S.; van der Gulden, H.; van der Heijden, I.; O’Brien, S.W.; et al. The BRCA1-Δ11q Alternative Splice Isoform Bypasses Germline Mutations and Promotes Therapeutic Resistance to PARP Inhibition and Cisplatin. Cancer Res. 2016, 76, 2778–2790.
- Cruz, C.; Castroviejo-Bermejo, M.; Gutiérrez-Enríquez, S.; Llop-Guevara, A.; Ibrahim, Y.H.; Gris-Oliver, A.; Bonache, S.; Morancho, B.; Bruna, A.; Rueda, O.M.; et al. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated breast cancer. Ann. Oncol. 2018, 29, 1203–1210.
- Wang, Y.; Bernhardy, A.J.; Nacson, J.; Krais, J.J.; Tan, Y.F.; Nicolas, E.; Radke, M.R.; Handorf, E.; Llop-Guevara, A.; Balmaña, J.; et al. BRCA1 intronic Alu elements drive gene rearrangements and PARP inhibitor resistance. Nat. Commun. 2019, 10, 5661.
- Castroviejo-Bermejo, M.; Cruz, C.; Llop-Guevara, A.; Gutiérrez-Enríquez, S.; Ducy, M.; Ibrahim, Y.H.; Gris-Oliver, A.; Pellegrino, B.; Bruna, A.; Guzmán, M.; et al. A RAD51 assay feasible in routine tumor samples calls PARP inhibitor response beyond BRCA mutation. EMBO Mol. Med. 2018, 10, e9172.
- Ter Brugge, P.; Kristel, P.; van der Burg, E.; Boon, U.; de Maaker, M.; Lips, E.; Mulder, L.; de Ruiter, J.; Moutinho, C.; Gevensleben, H.; et al. Mechanisms of Therapy Resistance in Patient-Derived Xenograft Models of BRCA1-Deficient Breast Cancer. J. Natl. Cancer Inst. 2016, 108, djw148, Erratum in J. Natl. Cancer Inst. 2020, 112, 1075.
- Veeck, J.; Ropero, S.; Setien, F.; Gonzalez-Suarez, E.; Osorio, A.; Benitez, J.; Herman, J.G.; Esteller, M. BRCA1 CpG island hypermethylation predicts sensitivity to poly(adenosine diphosphate)-ribose polymerase inhibitors. J. Clin. Oncol. 2010, 28, e563–e564, author reply e565-6.
- Wang, Y.Q.; Zhang, J.R.; Li, S.D.; He, Y.Y.; Yang, Y.X.; Liu, X.L.; Wan, X.P. Aberrant methylation of breast and ovarian cancer susceptibility gene 1 in chemosensitive human ovarian cancer cells does not involve the phosphatidylinositol 30-kinase-Akt pathway. Cancer Sci. 2010, 101, 1618–1623.
- Belotserkovskaya, R.; Raga Gil, E.; Lawrence, N.; Butler, R.; Clifford, G.; Wilson, M.D.; Jackson, S.P. PALB2 chromatin recruitment restores homologous recombination in BRCA1-deficient cells depleted of 53BP1. Nat. Commun. 2020, 11, 819.
- Xu, G.; Chapman, J.R.; Brandsma, I.; Yuan, J.; Mistrik, M.; Bouwman, P.; Bartkova, J.; Gogola, E.; Warmerdam, D.; Barazas, M.; et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 2015, 521, 541–544.
- Noordermeer, S.M.; Adam, S.; Setiaputra, D.; Barazas, M.; Pettitt, S.J.; Ling, A.K.; Olivieri, M.; Álvarez-Quilón, A.; Moatti, N.; Zimmermann, M.; et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 2018, 560, 117–121.
- Gupta, R.; Somyajit, K.; Narita, T.; Maskey, E.; Stanlie, A.; Kremer, M.; Typas, D.; Lammers, M.; Mailand, N.; Nussenzweig, A.; et al. DNA Repair Network Analysis Reveals Shieldin as a Key Regulator of NHEJ and PARP Inhibitor Sensitivity. Cell 2018, 173, 972–988.e23.
- He, Y.J.; Meghani, K.; Caron, M.C.; Yang, C.; Ronato, D.A.; Bian, J.; Sharma, A.; Moore, J.; Niraj, J.; Detappe, A.; et al. DYNLL1 binds to MRE11 to limit DNA end resection in BRCA1-deficient cells. Nature 2018, 563, 522–526.
- Kondrashova, O.; Nguyen, M.; Shield-Artin, K.; Tinker, A.V.; Teng, N.; Harrell, M.I.; Kuiper, M.J.; Ho, G.Y.; Barker, H.; Jasin, M.; et al. Secondary Somatic Mutations Restoring RAD51C and RAD51D Associated with Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2017, 7, 984–998.
- Liu, Y.; Burness, M.L.; Martin-Trevino, R.; Guy, J.; Bai, S.; Harouaka, R.; Brooks, M.D.; Shang, L.; Fox, A.; Luther, T.K.; et al. RAD51 Mediates Resistance of Cancer Stem Cells to PARP Inhibition in Triple-Negative Breast Cancer. Clin. Cancer Res. 2017, 23, 514–522.
- Marzio, A.; Puccini, J.; Kwon, Y.; Maverakis, N.K.; Arbini, A.; Sung, P.; Bar-Sagi, D.; Pagano, M. The F-Box Domain-Dependent Activity of EMI1 Regulates PARPi Sensitivity in Triple-Negative Breast Cancers. Mol. Cell 2019, 73, 224–237.e6.
- Michl, J.; Zimmer, J.; Buffa, F.M.; McDermott, U.; Tarsounas, M. FANCD2 limits replication stress and genome instability in cells lacking BRCA2. Nat. Struct. Mol. Biol. 2016, 23, 755–757.
- Chaudhuri, A.R.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387.
- Taglialatela, A.; Alvarez, S.; Leuzzi, G.; Sannino, V.; Ranjha, L.; Huang, J.W.; Madubata, C.; Anand, R.; Levy, B.; Rabadan, R.; et al. Restoration of replication fork stability in BRCA1-and BRCA2-deficient cells by inactivation of SNF2-family fork remodelers. Mol. Cell 2017, 68, 414–430.
- Dungrawala, H.; Bhat, K.P.; Le Meur, R.; Chazin, W.J.; Ding, X.; Sharan, S.K.; Wessel, S.R.; Sathe, A.A.; Zhao, R.; Cortez, D. RADX promotes genome stability and modulates chemosensitivity by regulating RAD51 at replication forks. Mol. Cell 2017, 67, 374–386.e5.
- Carnero, A.; Blanco-Aparicio, C.; Renner, O.; Link, W.; Leal, J.F. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Curr. Cancer Drug Targets 2008, 8, 187–198.
- Carnero, A. The PKB/AKT pathway in cancer. Curr. Pharm. Des. 2010, 16, 34–44.
- Hollander, M.C.; Blumenthal, G.M.; Dennis, P.A. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat. Rev. 2011, 11, 289–301.
- Nelson, A.C.; Lyons, T.R.; Young, C.D.; Hansen, K.C.; Anderson, S.M.; Holt, J.T. AKT regulates BRCA1 stability in response to hormone signaling. Mol. Cell. Endocrinol. 2010, 319, 129–142.
- Kang, H.J.; Yi, Y.W.; Kim, H.J.; Hong, Y.B.; Seong, Y.S.; Bae, I. BRCA1 negatively regulates IGF-1 expression through an estrogen-responsive element-like site. Cell Death Dis. 2012, 3, e336.
- Xiang, T.; Ohashi, A.; Huang, Y.; Pandita, T.K.; Ludwig, T.; Powell, S.N.; Yang, Q. Negative Regulation of AKT Activation by BRCA1. Cancer Res. 2008, 68, 10040–10044.
- Saal, L.H.; Gruvberger-Saal, S.K.; Persson, C.; Lövgren, K.; Jumppanen, M.; Staaf, J.; Jönsson, G.; Pires, M.M.; Maurer, M.; Holm, K.; et al. Recurrent gross mutations of the PTEN tumor suppressor gene in breast cancers with deficient DSB repair. Nat. Genet. 2008, 40, 102–107.
- Foulkes, W.D. BRCA1—Sowing the seeds crooked in the furrow. Nat. Genet. 2008, 40, 8–9.
- Shen, W.H.; Balajee, A.S.; Wang, J.; Wu, H.; Eng, C.; Pandolfi, P.P.; Yin, Y. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 2007, 128, 157–170.
- McEllin, B.; Camacho, C.V.; Mukherjee, B.; Hahm, B.; Tomimatsu, N.; Bachoo, R.M.; Burma, S. PTEN loss compromises homologous recombination repair in astrocytes: Implications for glioblastoma therapy with temozolomide or poly(ADP-ribose) polymerase inhibitors. Cancer Res. 2010, 70, 5457–5464.
- Mendes-Pereira, A.M.; Martin, S.A.; Brough, R.; McCarthy, A.; Taylor, J.R.; Kim, J.S.; Waldman, T.; Lord, C.J.; Ashworth, A. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol. Med. 2009, 1, 315–322.
- Minami, A.; Nakanishi, A.; Ogura, Y.; Kitagishi, Y.; Matsuda, S. Connection between Tumor Suppressor BRCA1 and PTEN in Damaged DNA Repair. Front. Oncol. 2014, 4, 318.
- Chock, K.L.; Allison, J.M.; Shimizu, Y.; El Shamy, W.M. BRCA1-IRIS overexpression promotes cisplatin resistance in ovarian cancer cells. Cancer Res. 2010, 70, 8782–8791.
- Burstein, H.J. Novel agents and future directions for refractory breast cancer. Semin. Oncol. 2011, 38, S17–S24.
- Yap, T.A.; Kristeleit, R.; Michalarea, V.; Pettitt, S.J.; im, J.S.; Carreira, S.; Roda, D.; Miller, R.; Riisnaes, R.; Miranda, S.; et al. Phase I Trial of the PARP Inhibitor Olaparib and AKT Inhibitor Capivasertib in Patients with BRCA1/2- and Non-BRCA1/2-Mutant Cancers. Cancer Discov. 2020, 10, 1528–1543.
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair pathway choices and consequences at the double-strand break. Trends Cell. Biol. 2016, 26, 52–64.
- Myler, L.R.; Gallardo, I.F.; Soniat, M.M.; Deshpande, R.A.; Gonzalez, X.B.; Kim, Y.; Paull, T.T.; Finkelstein, I.J. Single-Molecule Imaging Reveals How Mre11-Rad50-Nbs1 Initiates DNA Break Repair. Mol. Cell 2017, 67, 891–898.e4.
- Buis, J.; Stoneham, T.; Spehalski, E.; Ferguson, D.O. Mre11 regulates CtIP-dependent double-strand break repair by interaction with CDK2. Nat. Struct. Mol. Biol. 2012, 19, 246–252.
- Lee, J.H.; Paull, T.T. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 2004, 304, 93–96.
- Kumagai, A.; Lee, J.; Yoo, H.Y.; Dunphy, W.G. TopBP1 activates the ATR-ATRIP complex. Cell 2006, 124, 943–955.
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166.
- Ducy, M.; Sesma-Sanz, L.; Guitton-Sert, L.; Lashgari, A.; Gao, Y.; Brahiti, N.; Rodrigue, A.; Margaillan, G.; Caron, M.C.; Côté, J.; et al. The Tumor Suppressor PALB2: Inside Out. Trends Biochem. Sci. 2019, 44, 226–240.
- Sun, Y.; McCorvie, T.J.; Yates, L.A.; Zhang, X. Structural basis of homologous recombination. Cell. Mol. Life Sci. 2020, 77, 3–18.
- Cortesi, L.; Piombino, C.; Toss, A. Germline Mutations in Other Homologous Recombination Repair-Related Genes Than BRCA1/2: Predictive or Prognostic Factors? J. Pers. Med. 2021, 11, 245.
- Symington, L.S. Mechanism and regulation of DNA end resection in eukaryotes. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 195–212.
- Jowsey, P.; Morrice, N.A.; Hastie, C.J.; McLauchlan, H.; Toth, R.; Rouse, J. Characterisation of the sites of DNA damage-induced 53BP1 phosphorylation catalysed by ATM and ATR. DNA Repair 2007, 6, 1536–1544.
- Zhang, F.; Gong, Z. Regulation of DNA double-strand break repair pathway choice: A new focus on 53BP1. J. Zhejiang Univ. Sci. B 2021, 22, 38–46.
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The trinity at the heart of the DNA damage response. Mol. Cell 2017, 66, 801–817.
- Daley, J.M.; Sung, P. 53BP1, BRCA1 and the choice between recombination and end joining at DNA double-strand breaks. Mol. Cell. Biol. 2014, 34, 1380–1388.
- Feng, L.; Li, N.; Li, Y.; Wang, J.; Gao, M.; Wang, W.; Chen, J. Cell cycle-dependent inhibition of 53BP1 signaling by BRCA1. Cell Discov. 2015, 1, 15019.
- Hustedt, N.; Durocher, D. The control of DNA repair by the cell cycle. Nat. Cell Biol. 2017, 19, 1–9.
- Chanut, P.; Britton, S.; Coates, J.; Jackson, S.P.; Calsou, P. Coordinated nuclease activities counteract Ku at single-ended DNA double-strand breaks. Nat. Commun. 2016, 7, 12889.
- Ahel, D.; Hořejší, Z.; Wiechens, N.; Polo, S.E.; Garcia-Wilson, E.; Ahel, I.; Flynn, H.; Skehel, M.; West, S.C.; Jackson, S.P.; et al. Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science 2009, 325, 1240–1243.
- Gottschalk, A.J.; Timinszky, G.; Kong, S.E.; Jin, J.; Cai, Y.; Swanson, S.K.; Washburn, M.P.; Florens, L.; Ladurner, A.G.; Conaway, J.W.; et al. Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler. Proc. Natl. Acad. Sci. USA 2009, 106, 13770–13774.
- Ma, N.F.; Hu, L.; Fung, J.M.; Xie, D.; Zheng, B.J.; Chen, L.; Tang, D.J.; Fu, L.; Wu, Z.; Chen, M.; et al. Isolation and characterization of a novel oncogene, amplified in liver cancer 1, within a commonly amplified region at 1q21 in hepatocellular carcinoma. Hepatology 2008, 47, 503–510.
- Dagogo-Jack, I.; Shaw, A. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94.
- Kathawala, R.J.; Gupta., P.; Ashby, C.R., Jr.; Chen, Z.S. The modulation of ABC transporter-mediated multidrug resistance in cancer: A review of the past decade. Drug Resist. Updat. 2015, 18, 1–17.
- Shen, D.W.; Fojo, A.; Chin, J.E.; Roninson, I.B.; Richert, N.; Pastan, I.; Gottesman, M.M. Human multidrug-resistant cell lines: Increased mdr1 expression can precede gene amplification. Science 1986, 232, 643–645.
- Fojo, A.T.; Ueda, K.; Slamon, D.J.; Poplack, D.G.; Gottesman, M.M.; Pastan, I. Expression of a multidrug-resistance gene in human tumors and tissues. Proc. Natl. Acad. Sci. USA 1987, 84, 265–269.
- Parekh, H.; Wiesen, K.; Simpkins, H. Acquisition of taxol resistance via P-glycoprotein- and non-P-glycoprotein-mediated mechanisms in human ovarian carcinoma cells. Biochem. Pharmacol. 1997, 53, 461–470.
- Wang, Y.C.; Juric, D.; Francisco, B.; Yu, R.X.; Duran, G.E.; Chen, G.K.; Chen, X.; Sikic, B.I. Regional activation of chromosomal arm 7q with and without gene amplification in taxane-selected human ovarian cancer cell lines. Genes Chromosomes Cancer 2006, 45, 365–374.
- Rottenberg, S.; Jaspers, J.E.; Kersbergen, A.; van der Burg, E.; Nygren, A.O.; Zander, S.A.; Derksen, P.W.; de Bruin, M.; Zevenhoven, J.; Lau, A.; et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc. Natl. Acad. Sci. USA 2008, 105, 17079–17084.
- Wils, P.; Phung-Ba, V.; Warnery, A.; Lechardeur, D.; Raeissi, S.; Hidalgo, I.J.; Scherman, D. Polarized transport of docetaxel and vinblastine mediated by P-glycoprotein in human intestinal epithelial cell monolayers. Biochem. Pharmacol. 1994, 48, 1528–1530.
- Szakács, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234.
- Pascal, J.M.; Ellenberger, T. The rise and fall of poly(ADP-ribose): An enzymatic perspective. DNA Repair 2015, 32, 10–16.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.7K
Revisions:
2 times
(View History)
Update Date:
07 Jun 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No