 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jacinta Serpa | + 2486 word(s) | 2486 | 2020-10-06 11:23:29 | | | |
| 2 | Bruce Ren | + 15 word(s) | 2501 | 2020-10-09 08:23:33 | | | | |
| 3 | Karina Chen | Meta information modification | 2501 | 2020-10-26 09:28:13 | | |
Video Upload Options
Glutamine is a non-essential amino acid that plays a key role in the metabolism of proliferating cells including neoplastic cells. In the central nervous system (CNS), glutamine metabolism is particularly relevant, because the glutamine–glutamate cycle is a way of controlling the production of glutamate-derived neurotransmitters by tightly regulating the bioavailability of the amino acids in a neuron-astrocyte metabolic symbiosis-dependent manner. Glutamine-related metabolic adjustments have been reported in several CNS malignancies including malignant gliomas that are considered ‘glutamine addicted’. In these tumors, glutamine becomes an essential amino acid preferentially used in energy and biomass production including glutathione (GSH) generation, which is crucial in oxidative stress control. Therefore, in this review, we will highlight the metabolic remodeling that gliomas undergo, focusing on glutamine metabolism. We will address some therapeutic regimens including novel research attempts to target glutamine metabolism and a brief update of diagnosis strategies that take advantage of this altered profile. A better understanding of malignant glioma cell metabolism will help in the identification of new molecular targets and the design of new therapies.
1. Malignant Gliomas in Adult, an Overview
Central nervous system (CNS) tumors comprise a complex heterogeneous group of benign and malignant neoplasms from the brain and the spinal cord, having more than 100 histotypes [1][12][13][14][15][16][17][18][19][20][21][22][23][24][25][26][27][28][29][30][31][32][33][34][35][36][37][38][39][40][41][42][43][44][45][46][47][48][49][50][51][52][53][54][55][56][57][58][59][60][61][62][63][64][65][66][67][68][69][70][71][72][73][74][75][76][77][78][79][80][81][82][83][84][85]. The classification of CNS tumors was first based on histological variants, being classified according to their morphological features and similarities [2]. Gliomas are brain tumors with glial origin that account for approximately 25.5% of all primary CNS neoplasias, being 80.8% of all the malignant neoplasias affecting the CNS [3]. These neoplasms are characterized by a high mortality rate [3], mainly due to their inaccessible localization in the brain, the high proliferation rate, and infiltrative/invasive capacity [4]. This group embraces several histological entities according to morphological similarities between tumor cells and normal glial cells such as astrocytomas, oligodendrogliomas, and glioblastomas (GBM). Furthermore, these tumors were graded on a malignancy scale, from I to IV: grade I is associated with a better prognosis and lower anaplasia, whilst grade IV is applied to mitotically active neoplasms with the highest degree of anaplasia, being associated with very poor outcomes [2]. However, in 2016, the genetic basis of these tumors was clarified and molecular parameters were also taken into account for World Health Organization (WHO) glioma stratification [5]. Hence, the diagnosis and stratification of diffuse gliomas was facilitated by the recognition of isocitrate dehydrogenase 1/2 (IDH1/2) mutations and 1p/19q codeletion as principal biomarkers. Mutations in IDH1/2 occur in the majority of low grade gliomas and secondary GBM, being less frequent in primary GBM [6]. Presently, gliomas are grouped into five main molecular subgroups: GBM IDH-wild type, GBM IDH-mutant, astrocytoma IDH-wild type, astrocytoma IDH-mutant, oligodendroglioma IDH‑mutant, and 1p/19q-codeleted [5]. Most importantly, 1p/19q-codeleted and IDH-mutant tumors present a better clinical outcome, while GBM IDH‑wild type presents the worst prognosis, being the most common (50–60%) and the most lethal brain tumors [7][8]. However, it remains highly heterogenic, since it includes patients with a wide range of overall survival (OS), from 1 to 80 months (average OS of 15 months) [9][10][11]. Additional molecular biomarkers are therefore needed to better understand these neoplasms, devise better therapeutic strategies, and increase the accuracy of both glioma diagnosis and prognosis.
2. Glutamine and Glutamate Metabolism in the Central Nervous System (CNS)
Glutamine is a non-essential amino acid and the most abundant amino acid in the blood, representing around 20% of the total free amino acid pool [12]. Glutamine plays a role in maintaining pH homeostasis and interorgan nitrogen exchange via ammonia (NH3) transport between most proliferating cells, being consequently crucial in the progression of many cancers[13]. As a nitrogen source, glutamine is used as a substrate for nucleotide (purines, pyrimidines, and amino sugars) and nicotinamide adenine dinucleotide phosphate (NADPH) synthesis [14]. As a carbon source, glutamine supplies the tricarboxylic acid (TCA) cycle with oxaloacetate, α-ketoglutarate, and acetyl-CoA, thus being responsible for ATP and macromolecules synthesis, preferentially replacing glucose in certain tumors [15][16][17]. Furthermore, glutamine is a precursor of glutamate, which is necessary for the synthesis of non-essential amino acids and glutathione (GSH), the most important reactive oxygen species (ROS) scavenger and detoxifying agent [18][19].
Glutamate is the most abundant amino acid in the brain, typically present at a concentration of 10–12 μM. However, too much glutamate can be prejudicial, since high levels of glutamate can overstimulate the postsynaptic neurons, leading to CNS damage and causing disturbances such as seizures [20]. Thus, the imbalance of the neuron–glia interactions is extremely important in brain homeostasis.
In the last decades, the term “tripartite synapse” was purposed and a new astrocytes function was discovered: the regulation of glutamate levels. In the healthy brain, glutamine is used to synthesize glutamate, which, as mentioned before, is an excitatory neurotransmitter and a precursor of the main inhibitory neurotransmitter γ-aminobutyric acid (GABA). Since neurons lack pyruvate carboxylase (PC)[21], they are incapable of performing de novo synthesis of glutamate or GABA from glucose. Thus, astrocytes and neurons establish a metabolic crosstalk in which astrocytes synthesize glutamine through the glutamate–glutamine cycle that will be later be available to neurons [22]. Basically, astrocytes clear out the glutamate from the synaptic cleft, through the Glutamate Transporter 1 (GLT-1) and Glutamate Aspartate Transporter (GLAST) [23]. Then, the glutamine synthetase (GS) catalyzes the glutamate amidation reaction, generating glutamine. This glutamine is then released from astrocytes via SNAT3 (sodium-coupled neutral amino acid transporter 3), ASCT2 (alanine/serine/cysteine transporter 2), and other transporters, and is imported by presynaptic neurons through SNAT1 and SNAT2 [23]. Afterward, glutamine is hydrolyzed in the neurons by glutaminase (GLS) to glutamate and ammonia. Glutamate is then packed into the synaptic vesicles and sent to the synaptic cleft during neurotransmission. Finally, it is taken up again by the astrocytes [24] (Figure 1). Therefore, astrocytes have the pivotal function of removing glutamate from the synapse, mitigating glutamate-induced excitotoxicity.
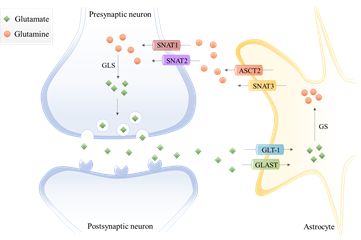
Figure 1. The glutamate-glutamine cycle in a glutamatergic synapse. The released neurotransmitter glutamate is imported by astrocytes through the glutamate transporter 1 (GLT-1) and glutamate aspartate transporter (GLAST). Then, glutamine synthetase (GS) catalyzes the glutamate amidation reaction, generating glutamine using free ammonia. The glutamine is then released from astrocytes via system A amino acid transporter 3 (SNAT3) and alanine/serine/cysteine-preferring transporter (ASCT2) and imported by presynaptic neurons through system A amino acid transporters 1 and 2 (SNAT1 and SNAT2). The glutamine is hydrolyzed to glutamate by glutaminase (GLS), which is packed into synaptic vesicles being sent to the synaptic cleft during neurotransmission. Finally, glutamate is imported again by the astrocytes.
Extracellular glutamate levels are also regulated by the cystine/glutamate antiporter xc–system. This glutamate transporter is predominantly expressed in astrocytes, oligodendrocytes, and in some cortical neurons [25]. The xc–system is pivotal in cell redox homeostasis, once it exchanges glutamate for cystine, which is converted to its reduced form cysteine [25]. This is one rate-limiting step of GSH synthesis, since it is a tripeptide composed of glutamate, cysteine, and glycine, with cysteine being the thiol component.
In the brain, glutamine synthetase (GS) is expressed in astrocytes, playing a crucial role in nitrogen metabolism [26]. Thus, astrocytes are responsible for ammonia detoxification [27] and the modulation of brain excitability [28] by participating in glutamate and GABA turnover.
Regarding glutaminase (GLS), the two isoforms (GLS-1 and GLS-2) are expressed in neurons and in astrocytes [29]. GLS-1 is the predominant GLS gene expressed in the brain and it encodes two splicing variants, the kidney-type glutaminase (KGA) and glutaminase C isoforms, with its expression modulated by oncogenes such as MYC [30], Rho GTPases [31], and Notch [32]. Through a surrogate promoter usage mechanism , GLS-2 encodes two liver-type isoforms, glutaminase 2 (GA) and liver-type glutaminase (LGA), and its expression can be regulated by p53 [33]. Glutaminase isoforms are activated upon low levels of phosphate , with GLS-2 being sensitive to lower phosphate concentrations than GLS-1. Furthermore, ammonia activates GLS-2 and inhibits GLS-1 .
3. Glutamine-Glutamate Relevance in Cancer
Cancer cells undergo metabolic alterations necessary for the acquisition of nutrients, essential for the production of biomass and energy that will sustain the high proliferative rate [34]. Glutamine catabolism is essential for mitochondrial metabolism, since glutamine provides anaplerotic carbons to supply the TCA cycle, accounting for ATP and macromolecules synthesis [35][36][37]. In cancer, glutamine is considered the main TCA cycle supplier upon cancer metabolic remodeling [38]. Increased glutaminolysis rate correlates with carcinogenesis, and its targeting impairs cancer cell proliferation [39][40][41].
The transporters capable of importing glutamine such as ATB0,+ (SLC6A14 gene), SNAT1 (SLC38A1 gene), ASCT2 (SLC1A5 gene), LAT1 (SLC7A5 gene), and LAT2 (SLC7A8 gene) [42] are crucial in cancer metabolic remodeling often being upregulated in tumors [43][44][45][46][47]. Therefore, glutamine transport targeting is currently being addressed in pre-clinical trials. LAT2 inhibition disturbs glutamine import and disrupts chemoresistance [46]. ASCT2 blockage impairs cancer metabolic remodeling, affecting cancer cell survival [47][48][49][50], thus specific inhibitors are under investigation for future clinical application [51][52]. However, the redundant activity of glutamine transporters [51][52] can be a mechanism of resistance to a glutamine uptake-targeted therapy.
In cytoplasm, glutamine is converted into glutamate (Figure 2) by glutaminase isoenzymes (GLS-1 and GLS-2) which are differently expressed amongst cancer types, presenting an overlapped metabolic function and being pointed as relevant modulators of the clinical outcomes [53]. GLS-2 presents a non-metabolic role related to p53 activation [54] and Snail transcription factor inhibition [55], accounting for GLS-2 classification as a tumor suppressor [55]. In malignant gliomas, GLS-2 is commonly downregulated, but GLS-1 is expressed [56][57], being pivotal in glutaminolysis, which in turn is crucial for GBM cell survival and tumor growth [58].
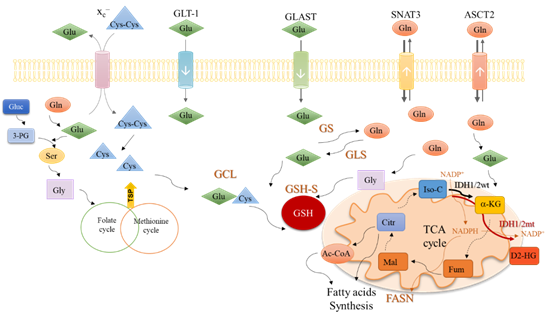
Figure 2. An integrative view of glutamine metabolism. Glutamine (Gln) is a core nutrient in cell metabolism. Gln can be synthesized within the cancer cell, by glutamine synthase (GS) or be taken up from the tumor microenvironment, though different transporters, as SNAT3 and ASCT2. Gln is catalyzed by glutaminases (GLS) enzymes and glutamate (Glu) is generated. Glu can be uptaken from the tumor microenvironment in a process mediated by transporters such as GLT-1 and GLAST. Glu controls the entrance of cystine (Cys-Cys) in the cell, mediated by xc– antiporter; Cys-Cys is then converted into cysteine (Cys). Gln-derived Glu is used as a nitrogen source in the synthesis of serine (Ser) from glucose (Gluc)-derived 3-phosphoglycerate (3-PG). Serine (Ser) can be converted into glycine (Gly) that supplies one-carbon metabolism (folate cycle plus methionine cycle) from which cysteine (Cys) is synthesized through the transsulfuration pathway (TSP). Glu, Cys and Gly are the three components of glutathione (GSH), whose synthesis occurs in two steps. In the first step, Glu and Cys are linked by glutamyl-cysteine ligase (GCL) and afterward, Gly is added to the dipeptide Glu-Cys by glutathione synthase (GSH-S). Gln-derived Glu can be converted into α-ketoglutarate (α-KG) and enter the tricarboxylic acids (TCA) cycle. α-KG can also be synthesized by isocitrate dehydrogenase 1/2 wild type (IDH1/2wt) enzymes from isocitrate (Iso-C), with the consumption of NADP+ and release of NADPH. NADPH is canalized to other metabolic pathways such as the fatty acid synthesis catalyzed by the fatty acids synthase (FASN). The isocitrate dehydrogenase 1/2 mutant (IDH1/2mt) enzymes further catalyze the conversion of α-KG into the onco-metabolite, 2-hydroxyglutarate (D2-HG), with NADPH consumption. Different Gln-derived TCA cycle intermediates such as fumarate (Fum), malate (Mal), citrate (Citr), and acetyl-CoA (Ac-CoA) can be deviated to supply fatty acid synthesis.
Glutamate can be converted into α-ketoglutarate through oxidative deamination by glutamate dehydrogenase 1 (GLDH1) in the mitochondria, or through transamination by amino acid-specific transaminases in the cytoplasm or mitochondria. Aside from α-ketoglutarate, the transamination produces nonessential amino acids such as serine (Figure 2) and aspartate. In cancer, the activation of transaminases is controlled by the MAPK pathway, the main regulator of glutamine metabolism, indicating that glutamine is an important precursor of other amino acids [59][61][62] and not only a supplier of the TCA cycle. Glutamine-derived glutamate is the amino group donor in the serine synthesis pathway, in which glucose-derived 3-phosphoglycerate is subject to a step-wise sequence of reactions to give rise to serine [60]. Then, serine can be converted into glycine, under the action of serine hydroxymethyltransferase (SHMT). Next, glycine is canalized to the one-carbon metabolism (folate cycle plus methionine cycle), in which several organic compounds are generated and deviated to supply crucial mechanisms such as the epigenetic modulation (methyl and acetyl groups), nucleotides synthesis, anti-oxidant systems and amino acids, and lipid production (Figure 2) [61][62].
In the mitochondria, α-ketoglutarate enters the TCA cycle and originates other organic compounds such as fumarate, malate, and citrate [63][64] under the action of different enzymes, which are deregulated in cancer. For instance, the malic isoenzymes (ME1 and ME2) are upregulated in different cancer types [65][66][67][68], being observed as pro-tumorigenic [69] and suitable therapeutic targets . Interestingly, a direct correlation between ME1 and the pentose phosphate pathway (PPP) was shown, proving that ME1 forms a hetero-oligomer with 6-phosphogluconate dehydrogenase (6PGD), the limiting PPP enzyme, and increases its affinity to the substrate 6-phosphogluconate [70], prompting the deviation of glycolysis intermediates to PPP and promoting the TCA cycle reliance on glutamine. This way, glucose is mainly metabolized in biosynthetic pathways, leaving the bioenergetics role to be played by glutamine. Moreover, citrate synthase (CS) expression is upregulated in cancer upon metabolic stressful conditions such as hypoxia, favoring the metabolic glutamine reliance [71]. Glutamine is also important in biomass production through the above-mentioned intervention in amino acid synthesis and also as a source for lipids, since citrate and glutamine-originated acetyl-CoA is the most relevant lipid precursor [72][73], accounting for about 20% of lipogenic acetyl-CoA [72].
The reliance on glutamine metabolism presented by cancer cells can be altered in the GBM-IDH mutant, since IDH1/2 mutated enzymes use α-ketoglutarate as a substrate to produce the oncometabolite 2-hydroxyglutarate (D2-HG), therefore a higher commitment of glutamine-derived glutamate is needed to produce α-ketoglutarate that will not be used in the TCA cycle, as will be depicted later on in this review.
3.1. Glutamate-Glutamine Metabolic Remodeling: How Do Gliomas Profit?
In malignant gliomas, the glutamine metabolic remodeling is characterized by the abrogation of GS [74] and the increased expression of glutamine transporters such as ASCT2 [75] and SNAT3 [76]. Therefore, these neoplasms present an increased dependence on the import of glutamine upon the incapacity of producing it [77], acting as ‘glutamine traps’ by importing glutamine from the tumor microenvironment [78].
On the other hand, GLT-1 expression is decreased in glioma cells, impairing the glutamate uptake [79]. Therefore, the increased glutamine uptake and the decreased consumption of glutamate canalizes the excessive glutamate produced by the cell to GSH production and also to the import of cyst(e)ine through xc–. This phenomenon functions as a mechanism of resistance to radio- and chemotherapy, thereby promoting tumor cell survival [80][81][82][83][84][85] and the increased expression of glutamine transporters such as ASCT2 [75] and SNAT3 [76]. Therefore. Increased GSH levels are correlated with treatment resistance not only in gliomas [82], but also in other types of cancer [83]. Moreover, in a study with primary GBM patients performed by Takeuchi et al., strong xCT expression was associated with shorter progression-free survival (PFS) and OS, suggesting a possible role as a predictive factor in GBM [84].
Regarding GLS, low expression of GLS-2 isoforms is a feature of many brain tumors including GBM, anaplastic astrocytomas, and ependymomas [85]. Nevertheless, these tumors express significant amounts of GLS-1 [85]. These facts show that malignant glioma cells are able to fully metabolize glutamine, pointing out this amino acid metabolic route as a core pathway, and consequently a putative target in cancer.
The article has been published on 10.3390/biom10101370
References
- • Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. 。
- • Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO Classification of Tumours of the Central Nervous System. Acta Neuropathol. 2007, 114, 97–109. 。
- • Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro Oncol. 2019, 21, v1–v100. 。
- • Obara-Michlewska, M.; Szeliga, M. Targeting Glutamine Addiction in Gliomas. Cancers 2020, 12, 310.
- • Chen, R.; Smith-Cohn, M.; Cohen, A.L.; Colman, H. Glioma Subclassifications and Their Clinical Significance. Neurotherapeuttics 2017, 14, 284–297.
- • Cohen, A.L.; Holmen, S.L.; Colman, H. IDH1 and IDH2 Mutations in Gliomas. Curr. Neurol. Neurosci. Rep. 2013, 13, 345.
- • Iuchi, T.; Sugiyama, T.; Ohira, M.; Kageyama, H.; Yokoi, S.; Sakaida, T.; Hasegawa, Y.; Setoguchi, T.; Itami, M. Clinical significance of the 2016 WHO classification in Japanese patients with gliomas. Brain Tumor Pathol. 2018, 35, 71–80.
- • Tabouret, E.; Network, F.P.; Nguyen, A.T.; Dehais, C.; Carpentier, C.; Ducray, F.; Idbaih, A.; Mokhtari, K.; Jouvet, A.; Uro-Coste, E.; et al. Prognostic impact of the 2016 WHO classification of diffuse gliomas in the French POLA cohort. Acta Neuropathol. 2016, 132, 625–634.
- • Brito, C.; Azevedo, A.; Esteves, S.; Marques, A.R.; Martins, C.; Costa, I.; Mafra, M.; Marques, J.M.B.; Roque, L.; Pojo, M. Clinical insights gained by refining the 2016 WHO classification of diffuse gliomas with: EGFR amplification, TERT mutations, PTEN deletion and MGMT methylation. BMC Cancer 2019, 19, 968.
- • Stupp, R.; Mason, W.P.; Bent, M.J.V.D.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996.
- • Batich, K.A.; Reap, E.A.; Archer, G.E.; Sanchez-Perez, L.; Nair, S.K.; Schmittling, R.J.; Norberg, P.; Xie, W.; Herndon, J.E.; Healy, P.; et al. Long-term Survival in Glioblastoma with Cytomegalovirus pp65-Targeted Vaccination. Clin. Cancer Res. 2017, 23, 1898–1909. 。
- • Cruzat, V.; Rogero, M.M.; Keane, K.N.; Curi, R.; Newsholme, P. Glutamine: Metabolism and Immune Function, Supplementation and Clinical Translation. Nutrients 2018, 10, 1564. 。
- • Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer 2017, 3, 169–180. 。
- • Curi, R.; Lagranha, C.J.; Doi, S.Q.; Sellitti, D.F.; Procopio, J.; Pithon-Curi, T.C.; Corless, M.; Newsholme, P. Molecular mechanisms of glutamine action. J. Cell. Physiol. 2005, 204, 392–401.
- • DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350.
- • Serpa, J. Metabolic Remodeling as a Way of Adapting to Tumor Microenvironment (TME), a Job of Several Holders. In Tumor Microenvironent—Main Driver Metabolic Adaptation; Serpa, J., Ed.; Springer Nature: Cham, Switzerland, 2020; pp. 1–34.
- • Lopes-Coelho, F.; Gouveia-Fernandes, S.; Gonçalves, L.G.; Nunes, C.; Faustino, I.; Silva, F.; Felix, A.; Pereira, S.; Serpa, J. HNF1β drives glutathione (GSH) synthesis underlying intrinsic carboplatin resistance of ovarian clear cell carcinoma (OCCC). Tumor Biol. 2015, 37, 4813–4829.
- • Santos, I.; Ramos, C.; Mendes, C.; Sequeira, C.O.; Tomé, C.S.; Fernandes, D.G.H.; Mota, P.; Pires, R.F.; Urso, D.; Hipólito, A.; et al. Targeting Glutathione and Cystathionine β-Synthase in Ovarian Cancer Treatment by Selenium-Chrysin Polyurea Dendrimer Nanoformulation. Nutrients 2019, 11, 2523.
- • Nunes, S.C.; Serpa, J. Glutathione in Ovarian Cancer: A Double-Edged Sword. Int. J. Mol. Sci. 2018, 19, 1882.
- • Gross, L. “Supporting” players take the lead in protecting the overstimulated brain. PLoS Biol. 2006, 4, e371.
- • Yu, A.C.H.; Drejer, J.; Hertz, L.; Schousboe, A. Pyruvate Carboxylase Activity in Primary Cultures of Astrocytes and Neurons. J. Neurochem. 1983, 41, 1484–1487.
- • Cooper, A.J.L.; Jeitner, T.M. Central Role of Glutamate Metabolism in the Maintenance of Nitrogen Homeostasis in Normal and Hyperammonemic Brain. Biomolecules 2016, 6, 16.
- • Bak, L.K.; Schousboe, A.; Waagepetersen, H.S. The glutamate/GABA-glutamine cycle: Aspects of transport, neurotransmitter homeostasis and ammonia transfer. J. Neurochem. 2006, 98, 641–653. 。
- • Mckenna, M.C.; Ferreira, G.C. The Glutamate/GABA-Glutamine Cycle. Neurodegener. Dis. 2016, 13, 59–98.
- • Burdo, J.; Dargusch, R.; Schubert, D. Distribution of the Cystine/Glutamate Antiporter System x−cin the Brain, Kidney, and Duodenum. J. Histochem. Cytochem. 2006, 54, 549–557. 。
- • Martinez-Hernandez, A.; Bell, K.; Norenberg, M. Glutamine synthetase: Glial localization in brain. Science 1977, 195, 1356–1358.
- • Cooper, A.J.L. The Role of Glutamine Synthetase and Glutamate Dehydrogenase in Cerebral Ammonia Homeostasis. Neurochem. Res. 2012, 37, 2439–2455.
- • Bellot-Saez, A.; Kékesi, O.; Morley, J.W.; Buskila, Y. Astrocytic modulation of neuronal excitability through K + spatial buffering. Neurosci. Biobehav. Rev. 2017, 77, 87–97.
- • Cardona, C.; Sanchez-Mejias, E.; Dávila, J.C.; Martín-Rufián, M.; Campos-Sandoval, J.A.; Vitorica, J.; Alonso, F.J.; Matés, J.M.; Segura, J.A.; Norenberg, M.D.; et al. Expression of Gls and Gls2 glutaminase isoforms in astrocytes. Glia 2014, 63, 365–382.
- • Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. C-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765.
- • Wang, J.-B.; Erickson, J.W.; Fuji, R.; Ramachandran, S.; Gao, P.; Dinavahi, R.; Wilson, K.F.; Ambrosio, A.L.; Dias, S.M.G.; Dang, C.V.; et al. Targeting Mitochondrial Glutaminase Activity Inhibits Oncogenic Transformation. Cancer Cell 2010, 18, 207–219.
- • Kahlert, U.D.; Cheng, M.; Koch, K.; Marchionni, L.; Fan, X.; Raabe, E.H.; Maciaczyk, J.; Glunde, K.; Eberhart, C.G. Alterations in cellular metabolome after pharmacological inhibition of Notch in glioblastoma cells. Int. J. Cancer 2016, 138, 1246–1255. 。
- • Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460.
- • Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2017, 23, 27–47. 。
- • Matés, J.M.; Segura, J.A.; Martin-Rufian, M.; Campos-Sandoval, J.A.; Alonso, F.J.; Marquez, J. Glutaminase Isoenzymes as Key Regulators in Metabolic and Oxidative Stress Against Cancer. Curr. Mol. Med. 2013, 13, 514–534. 。
- • Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. 。
- • Reynolds, M.R.; Lane, A.N.; Robertson, B.; Kemp, S.; Liu, Y.; Hill, B.G.; Dean, D.C.; Clem, B.F. Control of glutamine metabolism by the tumor suppressor Rb. Oncogene 2013, 33, 556–566.
- • Gaglio, D.; Metallo, C.M.; Gameiro, P.A.; Hiller, K.; Danna, L.S.; Balestrieri, C.; Alberghina, L.; Stephanopoulos, G.; Chiaradonna, F. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol. Syst. Biol. 2011, 7, 523.
- • Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.-Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787.
- • Lukey, M.J.; Wilson, K.F.; Cerione, R.A. Therapeutic strategies impacting cancer cell glutamine metabolism. Futur. Med. Chem. 2013, 5, 1685–1700.
- • Chen, L.; Cui, H. Targeting Glutamine Induces Apoptosis: A Cancer Therapy Approach. Int. J. Mol. Sci. 2015, 16, 22830–22855.
- • Bhutia, Y.D.; Ganapathy, V. Glutamine transporters in mammalian cells and their functions in physiology and cancer. Acta Mol. Cell Res. 2015, 1863, 2531–2539. 。
- • Ko, Y.-H.; Lin, Z.; Flomenberg, N.; Pestell, R.G.; Howell, A.; Sotgia, F.; Lisanti, M.P.; Martinez-Outschoorn, U.E. Glutamine fuels a vicious cycle of autophagy in the tumor stroma and oxidative mitochondrial metabolism in epithelial cancer cells. Cancer Biol. Ther. 2011, 12, 1085–1097. 。
- • Bothwell, P.; Kron, C.; Wittke, E.; Czerniak, B.; Bode, B. Targeted Suppression and Knockout of ASCT2 or LAT1 in Epithelial and Mesenchymal Human Liver Cancer Cells Fail to Inhibit Growth. Int. J. Mol. Sci. 2018, 19, 2093. 。
- • Rajasinghe, L.; Hutchings, M.; Gupta, S. Delta-Tocotrienol Modulates Glutamine Dependence by Inhibiting ASCT2 and LAT1 Transporters in Non-Small Cell Lung Cancer (NSCLC) Cells: A Metabolomic Approach. Metabolites 2019, 9, 50.
- • Feng, M.; Xiong, G.; Cao, Z.; Yang, G.; Zheng, S.; Qiu, J.; You, L.; Zheng, L.; Zhang, T.; Zhao, Y. LAT2 regulates glutamine-dependent mTOR activation to promote glycolysis and chemoresistance in pancreatic cancer. J. Exp. Clin. Cancer Res. 2018, 37, 274.
- • Bolzoni, M.; Chiu, M.; Accardi, F.; Vescovini, R.; Airoldi, I.; Storti, P.; Todoerti, K.; Agnelli, L.; Missale, G.; Andreoli, R.; et al. Dependence on glutamine uptake and glutamine addiction characterize myeloma cells: A new attractive target. Blood 2016, 128, 667–679.
- • Wahi, K.; Holst, J. ASCT2: A potential cancer drug target. Expert Opin. Ther. Targets 2019, 23, 555–558.
- • Giuliani, N.; Chiu, M.; Bolzoni, M.; Accardi, F.; Bianchi, M.G.; Toscani, D.; Aversa, F.; Bussolati, O. The potential of inhibiting glutamine uptake as a therapeutic target for multiple myeloma. Expert Opin. Ther. Targets 2017, 21, 231–234.
- • Wang, Q.; Beaumont, K.A.; Otte, N.J.; Font, J.; Bailey, C.G.; van Geldermalsen, M.; Sharp, D.M.; Tiffen, J.C.; Ryan, R.M.; Jormakka, M.; et al. Targeting glutamine transport to suppress melanoma cell growth. Int. J. Cancer 2014, 135, 1060–1071.
- • Bröer, A.; Gauthier-Coles, G.; Rahimi, F.; van Geldermalsen, M.; Dorsch, D.; Wegener, A.; Holst, J.; Bröer, S. Ablation of the ASCT2 (SLC1A5) gene encoding a neutral amino acid transporter reveals transporter plasticity and redundancy in cancer cells. J. Biol. Chem. 2019, 294, 4012–4026.
- • Bröer, A.; Fairweather, S.; Bröer, S. Disruption of Amino Acid Homeostasis by Novel ASCT2 Inhibitors Involves Multiple Targets. Front. Pharmacol. 2018, 9. 。
- • Saha, S.K.; Islam, S.M.; Abdullah-Al-Wadud, M.; Islam, S.; Ali, F.; Park, K.S.; Islam, S. Multiomics Analysis Reveals that GLS and GLS2 Differentially Modulate the Clinical Outcomes of Cancer. J. Clin. Med. 2019, 8, 355. 。
- • Zhang, C.; Liu, J.; Zhao, Y.; Yue, X.; Zhu, Y.; Wang, X.; Wu, H.; Blanco, F.; Li, S.; Bhanot, G.; et al. Glutaminase 2 is a novel negative regulator of small GTPase Rac1 and mediates p53 function in suppressing metastasis. eLife 2016, 5, e10727. 。
- • Kuo, T.-C.; Chen, C.-K.; Hua, K.-T.; Yu, P.; Lee, W.-J.; Chen, M.-W.; Jeng, Y.-M.; Chien, M.-H.; Kuo, K.-T.; Hsiao, M.; et al. Glutaminase 2 stabilizes Dicer to repress Snail and metastasis in hepatocellular carcinoma cells. Cancer Lett. 2016, 383, 282–294. 。
- • Szeliga, M.; Sidoryk, M.; Matyja, E.; Kowalczyk, P.; Albrecht, J. Lack of expression of the liver-type glutaminase (LGA) mRNA in human malignant gliomas. Neurosci. Lett. 2005, 374, 171–173. 。
- • Szeliga, M.; Bogacińska-Karaś, M.; Kuźmicz, K.; Rola, R.; Albrecht, J. Downregulation ofGLS2in glioblastoma cells is related to DNA hypermethylation but not to the p53 status. Mol. Carcinog. 2015, 55, 1309–1316.
- • Mukherjee, P.; Augur, Z.M.; Li, M.; Hill, C.; Greenwood, B.; Domin, M.A.; Kondakci, G.; Narain, N.R.; Kiebish, M.A.; Bronson, R.T.; et al. Therapeutic benefit of combining calorie-restricted ketogenic diet and glutamine targeting in late-stage experimental glioblastoma. Commun. Biol. 2019, 2, 200. 。
- • Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105.
- • Mullarky, E.; Lairson, L.L.; Cantley, L.C.; Lyssiotis, C.A. A novel small-molecule inhibitor of 3-phosphoglycerate dehydrogenase. Mol. Cell. Oncol. 2016, 3, e1164280.
- • Ducker, G.S.; Ghergurovich, J.M.; Mainolfi, N.; Suri, V.; Jeong, S.K.; Li, S.H.-J.; Friedman, A.; Manfredi, M.G.; Gitai, Z.; Kim, H.; et al. Human SHMT inhibitors reveal defective glycine import as a targetable metabolic vulnerability of diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. USA 2017, 114, 11404–11409.
- • Marani, M.; Paone, A.; Fiascarelli, A.; Macone, A.; Gargano, M.; Rinaldo, S.; Giardina, G.; Pontecorvi, V.; Koes, D.; McDermott, L.; et al. A pyrazolopyran derivative preferentially inhibits the activity of human cytosolic serine hydroxymethyltransferase and induces cell death in lung cancer cells. Oncotarget 2016, 7, 4570–4583.
- • Le, A.; Lane, A.N.; Hamaker, M.; Bose, S.; Gouw, A.; Barbi, J.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Zhang, H.; et al. Glucose-Independent Glutamine Metabolism via TCA Cycling for Proliferation and Survival in B Cells. Cell Metab. 2012, 15, 110–121. 。
- • Li, J.; Li, X.; Wu, L.; Pei, M.; Li, H.; Jiang, Y. miR-145 inhibits glutamine metabolism through c-myc/GLS1 pathways in ovarian cancer cells. Cell Biol. Int. 2019, 43, 921–930. 。
- • Sarfraz, I.; Rasul, A.; Hussain, G.; Hussain, S.M.; Ahmad, M.; Nageen, B.; Jabeen, F.; Selamoglu, Z.; Ali, M. Malic enzyme 2 as a potential therapeutic drug target for cancer. IUBMB Life 2018, 70, 1076–1083. 。
- • Lu, Y.-X.; Ju, H.-Q.; Liu, Z.-X.; Chen, D.-L.; Wang, Y.; Zhao, Q.; Wu, Q.-N.; Zeng, Z.-L.; Qiu, H.-B.; Hu, P.-S.; et al. ME1 Regulates NADPH Homeostasis to Promote Gastric Cancer Growth and Metastasis. Cancer Res. 2018, 78, 1972–1985. 。
- • Liu, M.; Chen, Y.; Huang, B.; Mao, S.; Cai, K.; Wang, L.; Yao, X. Tumor-suppressing effects of microRNA-612 in bladder cancer cells by targeting malic enzyme 1 expression. Int. J. Oncol. 2018, 52, 1923–1933. 。
- • Nakashima, C.; Yamamoto, K.; Fujiwara-Tani, R.; Luo, Y.; Matsushima, S.; Fujii, K.; Ohmori, H.; Sasahira, T.; Sasaki, T.; Kitadai, Y.; et al. Expression of cytosolic malic enzyme (ME1) is associated with disease progression in human oral squamous cell carcinoma. Cancer Sci. 2018, 109, 2036–2045. 。
- • Fernandes, L.M.; Al-Dwairi, A.; Simmen, R.C.M.; Marji, M.; Brown, D.M.; Jewell, S.W.; Simmen, F.A. Malic Enzyme 1 (ME1) is pro-oncogenic in ApcMin/+ mice. Sci. Rep. 2018, 8, 14268.
- • Yao, P.; Sun, H.; Xu, C.; Chen, T.; Zou, B.; Jiang, P.; Du, W. Evidence for a direct cross-talk between malic enzyme and the pentose phosphate pathway via structural interactions. J. Biol. Chem. 2017, 292, 17113–17120.
- • Peng, M.; Yang, D.; Hou, Y.; Liu, S.; Zhao, M.; Qin, Y.; Chen, R.; Teng, Y.; Liu, M. Intracellular citrate accumulation by oxidized ATM-mediated metabolism reprogramming via PFKP and CS enhances hypoxic breast cancer cell invasion and metastasis. Cell Death Dis. 2019, 10, 228.
- • Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011, 481, 380–384. 。
- • Jiang, J.; Pavlova, N.N.; Zhang, J. Asparagine, a critical limiting metabolite during glutamine starvation. Mol. Cell. Oncol. 2018, 5, e1441633. 。
- • Castegna, A.; Menga, A. Glutamine synthetase: Localization dictates outcome. Genes 2018, 9, 108.
- • Kobayashi, M.; Mizutani, A.; Nishi, K.; Nakajima, S.; Shikano, N.; Nishii, R.; Fukuchi, K.; Kawai, K. Differences in accumulation and the transport mechanism of l- and d-methionine in high- and low-grade human glioma cells. Nucl. Med. Biol. 2017, 44, 78–82.
- • Sidoryk, M.; Matyja, E.; Dybel, A.; Zielińska, M.; Bogucki, J.; Jaskolski, D.J.; Liberski, P.P.; Kowalczyk, P.; Albrecht, J. Increased expression of a glutamine transporter SNAT3 is a marker of malignant gliomas. NeuroReport 2004, 15, 575–578. 。
- • Márquez, J.; Alonso, F.J.; Matés, J.M.; Segura, J.A.; Martín-Rufián, M.; Campos-Sandoval, J.A.; Márquez, J.D. Glutamine Addiction In Gliomas. Neurochem. Res. 2017, 42, 1735–1746. 。
- • Natarajan, S.K.; Venneti, S. Glutamine metabolism in brain tumors. Cancers 2019, 11, 1628. 。
- • Ye, Z.-C.; Rothstein, J.D.; Sontheimer, H. Compromised Glutamate Transport in Human Glioma Cells: Reduction–Mislocalization of Sodium-Dependent Glutamate Transporters and Enhanced Activity of Cystine–Glutamate Exchange. J. Neurosci. 1999, 19, 10767–10777.
- • McBrayer, S.K.; Mayers, J.R.; DiNatale, G.J.; Shi, D.D.; Khanal, J.; Chakraborty, A.A.; Sarosiek, K.; Briggs, K.J.; Robbins, A.K.; Sewastianik, T.; et al. Transaminase Inhibition by 2-Hydroxyglutarate Impairs Glutamate Biosynthesis and Redox Homeostasis in Glioma. Cell 2018, 175, 101–116.e25.
- • Shih, A.Y.; Erb, H.; Sun, X.; Toda, S.; Kalivas, P.W.; Murphy, T.H. Cystine/Glutamate Exchange Modulates Glutathione Supply for Neuroprotection from Oxidative Stress and Cell Proliferation. J. Neurosci. 2006, 26, 10514–10523.
- • Rocha, C.R.R.; Kajitani, G.S.; Quinet, A.; Fortunato, R.S.; Menck, C.F.M. NRF2 and glutathione are key resistance mediators to temozolomide in glioma and melanoma cells. Oncotarget 2016, 7. 。
- • Nunes, S.C.; Lopes-Coelho, F.; Gouveia-Fernandes, S.; Ramos, C.; Pereira, S.A.; Serpa, J. Cysteine boosters the evolutionary adaptation to CoCl2 mimicked hypoxia conditions, favouring carboplatin resistance in ovarian cancer. BMC Evol. Biol. 2018, 18, 1–17.
- • Takeuchi, S.; Wada, K.; Toyooka, T.; Shinomiya, N.; Shimazaki, H.; Nakanishi, K.; Nagatani, K.; Otani, N.; Osada, H.; Uozumi, Y.; et al. Increased xCT Expression Correlates With Tumor Invasion and Outcome in Patients with Glioblastomas. Neurosurgery 2013, 72, 33–41. 。
- • Szeliga, M.; Matyja, E.; Obara, M.; Grajkowska, W.; Czernicki, T.; Albrecht, J. Relative Expression of mRNAS Coding for Glutaminase Isoforms in CNS Tissues and CNS Tumors. Neurochem. Res. 2008, 33, 808–813.

