Glutamine is a non-essential amino acid that plays a key role in the metabolism of proliferating cells including neoplastic cells. In the central nervous system (CNS), glutamine metabolism is particularly relevant, because the glutamine–glutamate cycle is a way of controlling the production of glutamate-derived neurotransmitters by tightly regulating the bioavailability of the amino acids in a neuron-astrocyte metabolic symbiosis-dependent manner. Glutamine-related metabolic adjustments have been reported in several CNS malignancies including malignant gliomas that are considered ‘glutamine addicted’. In these tumors, glutamine becomes an essential amino acid preferentially used in energy and biomass production including glutathione (GSH) generation, which is crucial in oxidative stress control. Therefore, in this review, we will highlight the metabolic remodeling that gliomas undergo, focusing on glutamine metabolism. We will address some therapeutic regimens including novel research attempts to target glutamine metabolism and a brief update of diagnosis strategies that take advantage of this altered profile. A better understanding of malignant glioma cell metabolism will help in the identification of new molecular targets and the design of new therapies.
- malignant gliomas
- glutamine-glutamate cycle
- CNS
- cancer metabolism
- metabolic adaptation
- new metabolic-driven targets
1. Malignant Gliomas in Adult, an Overview
- Malignant Gliomas in Adult, an Overview
Central nervous system (CNS) tumors comprise a complex heterogeneous group of benign and malignant neoplasms from the brain and the spinal cord, having more than 100 histotypes [1][12][13][14][15][16][17][18][19][20][21][22][23][24][25][26][27][28][29][30][31][32][33][34][35][36][37][38][39][40][41][42][43][44][45][46][47][48][49][50][51][52][53][54][55][56][57][58][59][60][61][62][63][64][65][66][67][68][69][70][71][72][73][74][75][76][77][78][79][80][81][82][83][84][85]. The classification of CNS tumors was first based on histological variants, being classified according to their morphological features and similarities [2]. Gliomas are brain tumors with glial origin that account for approximately 25.5% of all primary CNS neoplasias, being 80.8% of all the malignant neoplasias affecting the CNS [3]. These neoplasms are characterized by a high mortality rate [3], mainly due to their inaccessible localization in the brain, the high proliferation rate, and infiltrative/invasive capacity [4]. This group embraces several histological entities according to morphological similarities between tumor cells and normal glial cells such as astrocytomas, oligodendrogliomas, and glioblastomas (GBM). Furthermore, these tumors were graded on a malignancy scale, from I to IV: grade I is associated with a better prognosis and lower anaplasia, whilst grade IV is applied to mitotically active neoplasms with the highest degree of anaplasia, being associated with very poor outcomes [2]. However, in 2016, the genetic basis of these tumors was clarified and molecular parameters were also taken into account for World Health Organization (WHO) glioma stratification [5]. Hence, the diagnosis and stratification of diffuse gliomas was facilitated by the recognition of isocitrate dehydrogenase 1/2 (
Central nervous system (CNS) tumors comprise a complex heterogeneous group of benign and malignant neoplasms from the brain and the spinal cord, having more than 100 histotypes [1]. The classification of CNS tumors was first based on histological variants, being classified according to their morphological features and similarities [2]. Gliomas are brain tumors with glial origin that account for approximately 25.5% of all primary CNS neoplasias, being 80.8% of all the malignant neoplasias affecting the CNS [3]. These neoplasms are characterized by a high mortality rate [3], mainly due to their inaccessible localization in the brain, the high proliferation rate, and infiltrative/invasive capacity [4]. This group embraces several histological entities according to morphological similarities between tumor cells and normal glial cells such as astrocytomas, oligodendrogliomas, and glioblastomas (GBM). Furthermore, these tumors were graded on a malignancy scale, from I to IV: grade I is associated with a better prognosis and lower anaplasia, whilst grade IV is applied to mitotically active neoplasms with the highest degree of anaplasia, being associated with very poor outcomes [2]. However, in 2016, the genetic basis of these tumors was clarified and molecular parameters were also taken into account for World Health Organization (WHO) glioma stratification [1,5]. Hence, the diagnosis and stratification of diffuse gliomas was facilitated by the recognition of isocitrate dehydrogenase 1/2 (
IDH1/2) mutations and 1p/19q codeletion as principal biomarkers. Mutations in
) mutations and 1p/19q codeletion as principal biomarkers [1]. Mutations in
IDH1/2 occur in the majority of low grade gliomas and secondary GBM, being less frequent in primary GBM [6]. Presently, gliomas are grouped into five main molecular subgroups: GBM
occur in the majority of low grade gliomas and secondary GBM, being less frequent in primary GBM [6]. Presently, gliomas are grouped into five main molecular subgroups: GBM
IDH
-wild type, GBM
IDH
-mutant, astrocytoma
IDH
-wild type, astrocytoma
IDH
-mutant, oligodendroglioma
IDH‑mutant, and 1p/19q-codeleted [5]. Most importantly, 1p/19q-codeleted and
‑mutant, and 1p/19q-codeleted [1,5]. Most importantly, 1p/19q-codeleted and
IDH
-mutant tumors present a better clinical outcome, while GBM
IDH‑wild type presents the worst prognosis, being the most common (50–60%) and the most lethal brain tumors [7][8]. However, it remains highly heterogenic, since it includes patients with a wide range of overall survival (OS), from 1 to 80 months (average OS of 15 months) [9][10][11]. Additional molecular biomarkers are therefore needed to better understand these neoplasms, devise better therapeutic strategies, and increase the accuracy of both glioma diagnosis and prognosis.
‑wild type presents the worst prognosis, being the most common (50–60%) and the most lethal brain tumors [7,8]. However, it remains highly heterogenic, since it includes patients with a wide range of overall survival (OS), from 1 to 80 months (average OS of 15 months) [9–11]. Additional molecular biomarkers are therefore needed to better understand these neoplasms, devise better therapeutic strategies, and increase the accuracy of both glioma diagnosis and prognosis.
2. Glutamine and Glutamate Metabolism in the Central Nervous System (CNS)
- Glutamine and Glutamate Metabolism in the Central Nervous System (CNS)
Glutamine is a non-essential amino acid and the most abundant amino acid in the blood, representing around 20% of the total free amino acid pool [12]. Glutamine plays a role in maintaining pH homeostasis and interorgan nitrogen exchange via ammonia (NH
3) transport between most proliferating cells, being consequently crucial in the progression of many cancers[13]. As a nitrogen source, glutamine is used as a substrate for nucleotide (purines, pyrimidines, and amino sugars) and nicotinamide adenine dinucleotide phosphate (NADPH) synthesis [14]. As a carbon source, glutamine supplies the tricarboxylic acid (TCA) cycle with oxaloacetate, α-ketoglutarate, and acetyl-CoA, thus being responsible for ATP and macromolecules synthesis, preferentially replacing glucose in certain tumors [15][16][17]. Furthermore, glutamine is a precursor of glutamate, which is necessary for the synthesis of non-essential amino acids and glutathione (GSH), the most important reactive oxygen species (ROS) scavenger and detoxifying agent [18][19].
) transport between most proliferating cells, being consequently crucial in the progression of many cancers [13]. As a nitrogen source, glutamine is used as a substrate for nucleotide (purines, pyrimidines, and amino sugars) and nicotinamide adenine dinucleotide phosphate (NADPH) synthesis [14]. As a carbon source, glutamine supplies the tricarboxylic acid (TCA) cycle with oxaloacetate, α-ketoglutarate, and acetyl-CoA, thus being responsible for ATP and macromolecules synthesis, preferentially replacing glucose in certain tumors [15–17]. Furthermore, glutamine is a precursor of glutamate, which is necessary for the synthesis of non-essential amino acids and glutathione (GSH), the most important reactive oxygen species (ROS) scavenger and detoxifying agent [18,19].
Glutamate is the most abundant amino acid in the brain, typically present at a concentration of 10–12 μM. However, too much glutamate can be prejudicial, since high levels of glutamate can overstimulate the postsynaptic neurons, leading to CNS damage and causing disturbances such as seizures [20]. Thus, the imbalance of the neuron–glia interactions is extremely important in brain homeostasis.
In the last decades, the term “tripartite synapse” was purposed and a new astrocytes function was discovered: the regulation of glutamate levels. In the healthy brain, glutamine is used to synthesize glutamate, which, as mentioned before, is an excitatory neurotransmitter and a precursor of the main inhibitory neurotransmitter γ-aminobutyric acid (GABA). Since neurons lack pyruvate carboxylase (PC)[21], they are incapable of performing de novo synthesis of glutamate or GABA from glucose. Thus, astrocytes and neurons establish a metabolic crosstalk in which astrocytes synthesize glutamine through the glutamate–glutamine cycle that will be later be available to neurons [22]. Basically, astrocytes clear out the glutamate from the synaptic cleft, through the Glutamate Transporter 1 (GLT-1) and Glutamate Aspartate Transporter (GLAST) [23]. Then, the glutamine synthetase (GS) catalyzes the glutamate amidation reaction, generating glutamine. This glutamine is then released from astrocytes via SNAT3 (sodium-coupled neutral amino acid transporter 3), ASCT2 (alanine/serine/cysteine transporter 2), and other transporters, and is imported by presynaptic neurons through SNAT1 and SNAT2 [23]. Afterward, glutamine is hydrolyzed in the neurons by glutaminase (GLS) to glutamate and ammonia. Glutamate is then packed into the synaptic vesicles and sent to the synaptic cleft during neurotransmission. Finally, it is taken up again by the astrocytes [24] (Figure 1). Therefore, astrocytes have the pivotal function of removing glutamate from the synapse, mitigating glutamate-induced excitotoxicity.
In the last decades, the term “tripartite synapse” was purposed and a new astrocytes function was discovered: the regulation of glutamate levels. In the healthy brain, glutamine is used to synthesize glutamate, which, as mentioned before, is an excitatory neurotransmitter and a precursor of the main inhibitory neurotransmitter γ-aminobutyric acid (GABA). Since neurons lack pyruvate carboxylase (PC) [21], they are incapable of performing de novo synthesis of glutamate or GABA from glucose. Thus, astrocytes and neurons establish a metabolic crosstalk in which astrocytes synthesize glutamine through the glutamate–glutamine cycle that will be later be available to neurons [22]. Basically, astrocytes clear out the glutamate from the synaptic cleft, through the Glutamate Transporter 1 (GLT-1) and Glutamate Aspartate Transporter (GLAST) [23]. Then, the glutamine synthetase (GS) catalyzes the glutamate amidation reaction, generating glutamine. This glutamine is then released from astrocytes via SNAT3 (sodium-coupled neutral amino acid transporter 3), ASCT2 (alanine/serine/cysteine transporter 2), and other transporters, and is imported by presynaptic neurons through SNAT1 and SNAT2 [23]. Afterward, glutamine is hydrolyzed in the neurons by glutaminase (GLS) to glutamate and ammonia. Glutamate is then packed into the synaptic vesicles and sent to the synaptic cleft during neurotransmission. Finally, it is taken up again by the astrocytes [4,24] (Figure 1). Therefore, astrocytes have the pivotal function of removing glutamate from the synapse, mitigating glutamate-induced excitotoxicity.
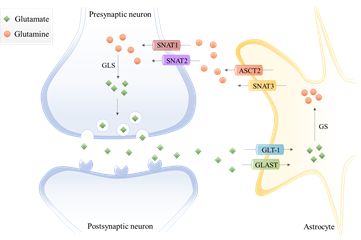
Figure 1.
The glutamate-glutamine cycle in a glutamatergic synapse. The released neurotransmitter glutamate is imported by astrocytes through the glutamate transporter 1 (GLT-1) and glutamate aspartate transporter (GLAST). Then, glutamine synthetase (GS) catalyzes the glutamate amidation reaction, generating glutamine using free ammonia. The glutamine is then released from astrocytes via system A amino acid transporter 3 (SNAT3) and alanine/serine/cysteine-preferring transporter (ASCT2) and imported by presynaptic neurons through system A amino acid transporters 1 and 2 (SNAT1 and SNAT2). The glutamine is hydrolyzed to glutamate by glutaminase (GLS), which is packed into synaptic vesicles being sent to the synaptic cleft during neurotransmission. Finally, glutamate is imported again by the astrocytes.
Extracellular glutamate levels are also regulated by the cystine/glutamate antiporter x
c–
system. This glutamate transporter is predominantly expressed in astrocytes, oligodendrocytes, and in some cortical neurons [25]. The x
c–system is pivotal in cell redox homeostasis, once it exchanges glutamate for cystine, which is converted to its reduced form cysteine [25]. This is one rate-limiting step of GSH synthesis, since it is a tripeptide composed of glutamate, cysteine, and glycine, with cysteine being the thiol component.
system is pivotal in cell redox homeostasis, once it exchanges glutamate for cystine, which is converted to its reduced form cysteine [18,25]. This is one rate-limiting step of GSH synthesis, since it is a tripeptide composed of glutamate, cysteine, and glycine, with cysteine being the thiol component [19].
In the brain, glutamine synthetase (GS) is expressed in astrocytes, playing a crucial role in nitrogen metabolism [26]. Thus, astrocytes are responsible for ammonia detoxification [27] and the modulation of brain excitability [28] by participating in glutamate and GABA turnover.
Regarding glutaminase (GLS), the two isoforms (GLS-1 and GLS-2) are expressed in neurons and in astrocytes [29]. GLS-1 is the predominant GLS gene expressed in the brain and it encodes two splicing variants, the kidney-type glutaminase (KGA) and glutaminase C isoforms, with its expression modulated by oncogenes such as MYC [30], Rho GTPases [31], and Notch [32]. Through a surrogate promoter usage mechanism , GLS-2 encodes two liver-type isoforms, glutaminase 2 (GA) and liver-type glutaminase (LGA), and its expression can be regulated by p53 [33]. Glutaminase isoforms are activated upon low levels of phosphate , with GLS-2 being sensitive to lower phosphate concentrations than GLS-1. Furthermore, ammonia activates GLS-2 and inhibits GLS-1 .
Regarding glutaminase (GLS), the two isoforms (GLS-1 and GLS-2) are expressed in neurons and in astrocytes [29]. GLS-1 is the predominant GLS gene expressed in the brain and it encodes two splicing variants, the kidney-type glutaminase (KGA) and glutaminase C isoforms, with its expression modulated by oncogenes such as MYC [30], Rho GTPases [31], and Notch [32]. Through a surrogate promoter usage mechanism [13], GLS-2 encodes two liver-type isoforms, glutaminase 2 (GA) and liver-type glutaminase (LGA), and its expression can be regulated by p53 [33]. Glutaminase isoforms are activated upon low levels of phosphate [23], with GLS-2 being sensitive to lower phosphate concentrations than GLS-1. Furthermore, ammonia activates GLS-2 and inhibits GLS-1 [4].
3. Glutamine-Glutamate Relevance in Cancer
- Glutamine-Glutamate Relevance in Cancer
Cancer cells undergo metabolic alterations necessary for the acquisition of nutrients, essential for the production of biomass and energy that will sustain the high proliferative rate [34]. Glutamine catabolism is essential for mitochondrial metabolism, since glutamine provides anaplerotic carbons to supply the TCA cycle, accounting for ATP and macromolecules synthesis [35][36][37]. In cancer, glutamine is considered the main TCA cycle supplier upon cancer metabolic remodeling [38]. Increased glutaminolysis rate correlates with carcinogenesis, and its targeting impairs cancer cell proliferation [39][40][41].
Cancer cells undergo metabolic alterations necessary for the acquisition of nutrients, essential for the production of biomass and energy that will sustain the high proliferative rate [34]. Glutamine catabolism is essential for mitochondrial metabolism, since glutamine provides anaplerotic carbons to supply the TCA cycle, accounting for ATP and macromolecules synthesis [35–37]. In cancer, glutamine is considered the main TCA cycle supplier upon cancer metabolic remodeling [38]. Increased glutaminolysis rate correlates with carcinogenesis, and its targeting impairs cancer cell proliferation [39–41].
The transporters capable of importing glutamine such as ATB
0,+
(
SLC6A14
gene), SNAT1 (
SLC38A1
gene), ASCT2 (
SLC1A5
gene), LAT1 (
SLC7A5
gene), and LAT2 (
SLC7A8 gene) [42] are crucial in cancer metabolic remodeling often being upregulated in tumors [43][44][45][46][47]. Therefore, glutamine transport targeting is currently being addressed in pre-clinical trials. LAT2 inhibition disturbs glutamine import and disrupts chemoresistance [46]. ASCT2 blockage impairs cancer metabolic remodeling, affecting cancer cell survival [47][48][49][50], thus specific inhibitors are under investigation for future clinical application [51][52]. However, the redundant activity of glutamine transporters [51][52] can be a mechanism of resistance to a glutamine uptake-targeted therapy.
gene) [42] are crucial in cancer metabolic remodeling often being upregulated in tumors [43–47]. Therefore, glutamine transport targeting is currently being addressed in pre-clinical trials. LAT2 inhibition disturbs glutamine import and disrupts chemoresistance [46]. ASCT2 blockage impairs cancer metabolic remodeling, affecting cancer cell survival [47–50], thus specific inhibitors are under investigation for future clinical application [51]. However, the redundant activity of glutamine transporters [51,52] can be a mechanism of resistance to a glutamine uptake-targeted therapy.
In cytoplasm, glutamine is converted into glutamate (Figure 2) by glutaminase isoenzymes (GLS-1 and GLS-2) which are differently expressed amongst cancer types, presenting an overlapped metabolic function and being pointed as relevant modulators of the clinical outcomes [53]. GLS-2 presents a non-metabolic role related to p53 activation [54] and Snail transcription factor inhibition [55], accounting for GLS-2 classification as a tumor suppressor [55]. In malignant gliomas, GLS-2 is commonly downregulated, but GLS-1 is expressed [56][57], being pivotal in glutaminolysis, which in turn is crucial for GBM cell survival and tumor growth [58].
In cytoplasm, glutamine is converted into glutamate (Figure 2) by glutaminase isoenzymes (GLS-1 and GLS-2) which are differently expressed amongst cancer types, presenting an overlapped metabolic function and being pointed as relevant modulators of the clinical outcomes [53]. GLS-2 presents a non-metabolic role related to p53 activation [54] and Snail transcription factor inhibition [55], accounting for GLS-2 classification as a tumor suppressor [53,55]. In malignant gliomas, GLS-2 is commonly downregulated, but GLS-1 is expressed [4,56,57], being pivotal in glutaminolysis, which in turn is crucial for GBM cell survival and tumor growth [58].
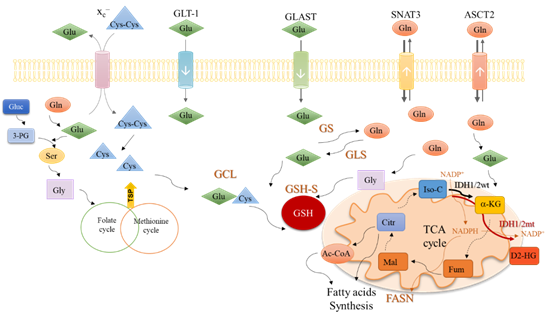
Figure 2.
An integrative view of glutamine metabolism. Glutamine (Gln) is a core nutrient in cell metabolism. Gln can be synthesized within the cancer cell, by glutamine synthase (GS) or be taken up from the tumor microenvironment, though different transporters, as SNAT3 and ASCT2. Gln is catalyzed by glutaminases (GLS) enzymes and glutamate (Glu) is generated. Glu can be uptaken from the tumor microenvironment in a process mediated by transporters such as GLT-1 and GLAST. Glu controls the entrance of cystine (Cys-Cys) in the cell, mediated by x
c–
antiporter; Cys-Cys is then converted into cysteine (Cys). Gln-derived Glu is used as a nitrogen source in the synthesis of serine (Ser) from glucose (Gluc)-derived 3-phosphoglycerate (3-PG). Serine (Ser) can be converted into glycine (Gly) that supplies one-carbon metabolism (folate cycle plus methionine cycle) from which cysteine (Cys) is synthesized through the transsulfuration pathway (TSP). Glu, Cys and Gly are the three components of glutathione (GSH), whose synthesis occurs in two steps. In the first step, Glu and Cys are linked by glutamyl-cysteine ligase (GCL) and afterward, Gly is added to the dipeptide Glu-Cys by glutathione synthase (GSH-S). Gln-derived Glu can be converted into α-ketoglutarate (α-KG) and enter the tricarboxylic acids (TCA) cycle. α-KG can also be synthesized by isocitrate dehydrogenase 1/2 wild type (IDH1/2wt) enzymes from isocitrate (Iso-C), with the consumption of NADP
+
and release of NADPH. NADPH is canalized to other metabolic pathways such as the fatty acid synthesis catalyzed by the fatty acids synthase (FASN). The isocitrate dehydrogenase 1/2 mutant (IDH1/2mt) enzymes further catalyze the conversion of α-KG into the onco-metabolite, 2-hydroxyglutarate (D2-HG), with NADPH consumption. Different Gln-derived TCA cycle intermediates such as fumarate (Fum), malate (Mal), citrate (Citr), and acetyl-CoA (Ac-CoA) can be deviated to supply fatty acid synthesis.
Glutamate can be converted into α-ketoglutarate through oxidative deamination by glutamate dehydrogenase 1 (GLDH1) in the mitochondria, or through transamination by amino acid-specific transaminases in the cytoplasm or mitochondria. Aside from α-ketoglutarate, the transamination produces nonessential amino acids such as serine (Figure 2) and aspartate. In cancer, the activation of transaminases is controlled by the MAPK pathway, the main regulator of glutamine metabolism, indicating that glutamine is an important precursor of other amino acids [59][61][62] and not only a supplier of the TCA cycle. Glutamine-derived glutamate is the amino group donor in the serine synthesis pathway, in which glucose-derived 3-phosphoglycerate is subject to a step-wise sequence of reactions to give rise to serine [60]. Then, serine can be converted into glycine, under the action of serine hydroxymethyltransferase (SHMT). Next, glycine is canalized to the one-carbon metabolism (folate cycle plus methionine cycle), in which several organic compounds are generated and deviated to supply crucial mechanisms such as the epigenetic modulation (methyl and acetyl groups), nucleotides synthesis, anti-oxidant systems and amino acids, and lipid production (Figure 2) [61][62].
Glutamate can be converted into α-ketoglutarate through oxidative deamination by glutamate dehydrogenase 1 (GLDH1) in the mitochondria, or through transamination by amino acid-specific transaminases in the cytoplasm or mitochondria. Aside from α-ketoglutarate, the transamination produces nonessential amino acids such as serine (Figure 2) and aspartate. In cancer, the activation of transaminases is controlled by the MAPK pathway, the main regulator of glutamine metabolism, indicating that glutamine is an important precursor of other amino acids [59] and not only a supplier of the TCA cycle. Glutamine-derived glutamate is the amino group donor in the serine synthesis pathway, in which glucose-derived 3-phosphoglycerate is subject to a step-wise sequence of reactions to give rise to serine [60]. Then, serine can be converted into glycine, under the action of serine hydroxymethyltransferase (SHMT). Next, glycine is canalized to the one-carbon metabolism (folate cycle plus methionine cycle), in which several organic compounds are generated and deviated to supply crucial mechanisms such as the epigenetic modulation (methyl and acetyl groups), nucleotides synthesis, anti-oxidant systems and amino acids, and lipid production (Figure 2) [61,62].
In the mitochondria, α-ketoglutarate enters the TCA cycle and originates other organic compounds such as fumarate, malate, and citrate [63][64] under the action of different enzymes, which are deregulated in cancer. For instance, the malic isoenzymes (ME1 and ME2) are upregulated in different cancer types [65][66][67][68], being observed as pro-tumorigenic [69] and suitable therapeutic targets . Interestingly, a direct correlation between ME1 and the pentose phosphate pathway (PPP) was shown, proving that ME1 forms a hetero-oligomer with 6-phosphogluconate dehydrogenase (6PGD), the limiting PPP enzyme, and increases its affinity to the substrate 6-phosphogluconate [70], prompting the deviation of glycolysis intermediates to PPP and promoting the TCA cycle reliance on glutamine. This way, glucose is mainly metabolized in biosynthetic pathways, leaving the bioenergetics role to be played by glutamine. Moreover, citrate synthase (CS) expression is upregulated in cancer upon metabolic stressful conditions such as hypoxia, favoring the metabolic glutamine reliance [71]. Glutamine is also important in biomass production through the above-mentioned intervention in amino acid synthesis and also as a source for lipids, since citrate and glutamine-originated acetyl-CoA is the most relevant lipid precursor [72][73], accounting for about 20% of lipogenic acetyl-CoA [72].
In the mitochondria, α-ketoglutarate enters the TCA cycle and originates other organic compounds such as fumarate, malate, and citrate [63,64] under the action of different enzymes, which are deregulated in cancer. For instance, the malic isoenzymes (ME1 and ME2) are upregulated in different cancer types [65–68], being observed as pro-tumorigenic [69] and suitable therapeutic targets [65,67]. Interestingly, a direct correlation between ME1 and the pentose phosphate pathway (PPP) was shown, proving that ME1 forms a hetero-oligomer with 6-phosphogluconate dehydrogenase (6PGD), the limiting PPP enzyme, and increases its affinity to the substrate 6-phosphogluconate [70], prompting the deviation of glycolysis intermediates to PPP and promoting the TCA cycle reliance on glutamine. This way, glucose is mainly metabolized in biosynthetic pathways, leaving the bioenergetics role to be played by glutamine. Moreover, citrate synthase (CS) expression is upregulated in cancer upon metabolic stressful conditions such as hypoxia, favoring the metabolic glutamine reliance [71]. Glutamine is also important in biomass production through the above-mentioned intervention in amino acid synthesis and also as a source for lipids, since citrate and glutamine-originated acetyl-CoA is the most relevant lipid precursor [63,72,73], accounting for about 20% of lipogenic acetyl-CoA [72].
The reliance on glutamine metabolism presented by cancer cells can be altered in the GBM-
IDH
mutant, since IDH1/2 mutated enzymes use α-ketoglutarate as a substrate to produce the oncometabolite 2-hydroxyglutarate (D2-HG), therefore a higher commitment of glutamine-derived glutamate is needed to produce α-ketoglutarate that will not be used in the TCA cycle, as will be depicted later on in this review.
3.1. Glutamate-Glutamine Metabolic Remodeling: How Do Gliomas Profit?
In malignant gliomas, the glutamine metabolic remodeling is characterized by the abrogation of GS [74] and the increased expression of glutamine transporters such as ASCT2 [75] and SNAT3 [76]. Therefore, these neoplasms present an increased dependence on the import of glutamine upon the incapacity of producing it [77], acting as ‘glutamine traps’ by importing glutamine from the tumor microenvironment [78].
In malignant gliomas, the glutamine metabolic remodeling is characterized by the abrogation of GS [74] and the increased expression of glutamine transporters such as ASCT2 [75] and SNAT3 [76]. Therefore, these neoplasms present an increased dependence on the import of glutamine upon the incapacity of producing it [77], acting as ‘glutamine traps’ by importing glutamine from the tumor microenvironment [78].
On the other hand, GLT-1 expression is decreased in glioma cells, impairing the glutamate uptake [79]. Therefore, the increased glutamine uptake and the decreased consumption of glutamate canalizes the excessive glutamate produced by the cell to GSH production and also to the import of cyst(e)ine through x
c–. This phenomenon functions as a mechanism of resistance to radio- and chemotherapy, thereby promoting tumor cell survival [80][81][82][83][84][85] and the increased expression of glutamine transporters such as ASCT2 [75] and SNAT3 [76]. Therefore. Increased GSH levels are correlated with treatment resistance not only in gliomas [82], but also in other types of cancer [83]. Moreover, in a study with primary GBM patients performed by Takeuchi et al., strong xCT expression was associated with shorter progression-free survival (PFS) and OS, suggesting a possible role as a predictive factor in GBM [84].
. This phenomenon functions as a mechanism of resistance to radio- and chemotherapy, thereby promoting tumor cell survival [80,81]. Increased GSH levels are correlated with treatment resistance not only in gliomas [82], but also in other types of cancer [19,83]. Moreover, in a study with primary GBM patients performed by Takeuchi et al., strong xCT expression was associated with shorter progression-free survival (PFS) and OS, suggesting a possible role as a predictive factor in GBM [84].
Regarding GLS, low expression of GLS-2 isoforms is a feature of many brain tumors including GBM, anaplastic astrocytomas, and ependymomas [85]. Nevertheless, these tumors express significant amounts of GLS-1 [85]. These facts show that malignant glioma cells are able to fully metabolize glutamine, pointing out this amino acid metabolic route as a core pathway, and consequently a putative target in cancer.
The article has been published on