Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Lihua Huang | -- | 3586 | 2022-06-04 16:05:17 | | | |
| 2 | Catherine Yang | Meta information modification | 3586 | 2022-06-06 04:40:29 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Huang, L.; , .; Hong-Min, L.; Zheng, Y. m6A Demethylases Inhibitors. Encyclopedia. Available online: https://encyclopedia.pub/entry/23724 (accessed on 24 July 2026).
Huang L, , Hong-Min L, Zheng Y. m6A Demethylases Inhibitors. Encyclopedia. Available at: https://encyclopedia.pub/entry/23724. Accessed July 24, 2026.
Huang, Lihua, , Liu Hong-Min, Yi-Chao Zheng. "m6A Demethylases Inhibitors" Encyclopedia, https://encyclopedia.pub/entry/23724 (accessed July 24, 2026).
Huang, L., , ., Hong-Min, L., & Zheng, Y. (2022, June 04). m6A Demethylases Inhibitors. In Encyclopedia. https://encyclopedia.pub/entry/23724
Huang, Lihua, et al. "m6A Demethylases Inhibitors." Encyclopedia. Web. 04 June, 2022.
Copy Citation
N6-methyladenosine (m6A) is a post-transcriptional RNA modification and one of the most abundant types of RNA chemical modifications. m6A functions as a molecular switch and is involved in a range of biomedical aspects, including cardiovascular diseases, the central nervous system, and cancers.
FTO
ALKBH5
human diseases
inhibitors
1. Introduction
Existing epigenetics mainly involve reversible chemical modifications of DNA, histone, and RNA, which can be inherited through cell division without changing the DNA sequence. So far, over 100 kinds of post-transcriptional modifications of RNA have been defined, including N1-methyladenosine (m1A), N6-methyladenosine (m6A), and 2-O-dimethyladenosine (m6Am) [1][2]. An important discovery was that m6A was the most abundant in RNA internal modifications between different species [1][3], and was also presented in eukaryotic mRNAs [4] and noncoding RNAs [5]. As with any other epigenetic modifications, m6A methylation can be individually dynamically installed; removed; and recognized by the so-called “writers”, “erasers”, and “readers”, individually (Figure 1) [6][7]. The occurrence of m6A methylation is controlled by a core methyltransferase complex, i.e., the “writers”, composed of several core proteins, including methyltransferase-like 3 and 14 (METTL3 and METTL14), and wilms tumor 1-associated protein (WTAP) [8]. Only two m6A demethylases “erasers”, fat-mass- and obesity-associated protein (FTO) [9] and alkB homolog 5 (ALKBH5) [10], have been found so far, which can specifically eliminate the m6A sites from target mRNAs. The “readers”, including YT521-B homology (YTH) domain family 1–3 (YTHDF1-3), YTH domain containing 1–2 (YTHDC1-2), insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs) (including IGF2BP1–3), and eukaryotic initiation factor 3 (EIF3), control the fate of the target mRNA by recognizing and binding to m6A sites [11]. Notably, these ‘readers’ perform different functions after binding to RNA containing m6A, resulting in different destinies for target RNA [12]. For example, YTHDF1 enhances RNA translation while YTHDF2 induces degradation of the transcripts.
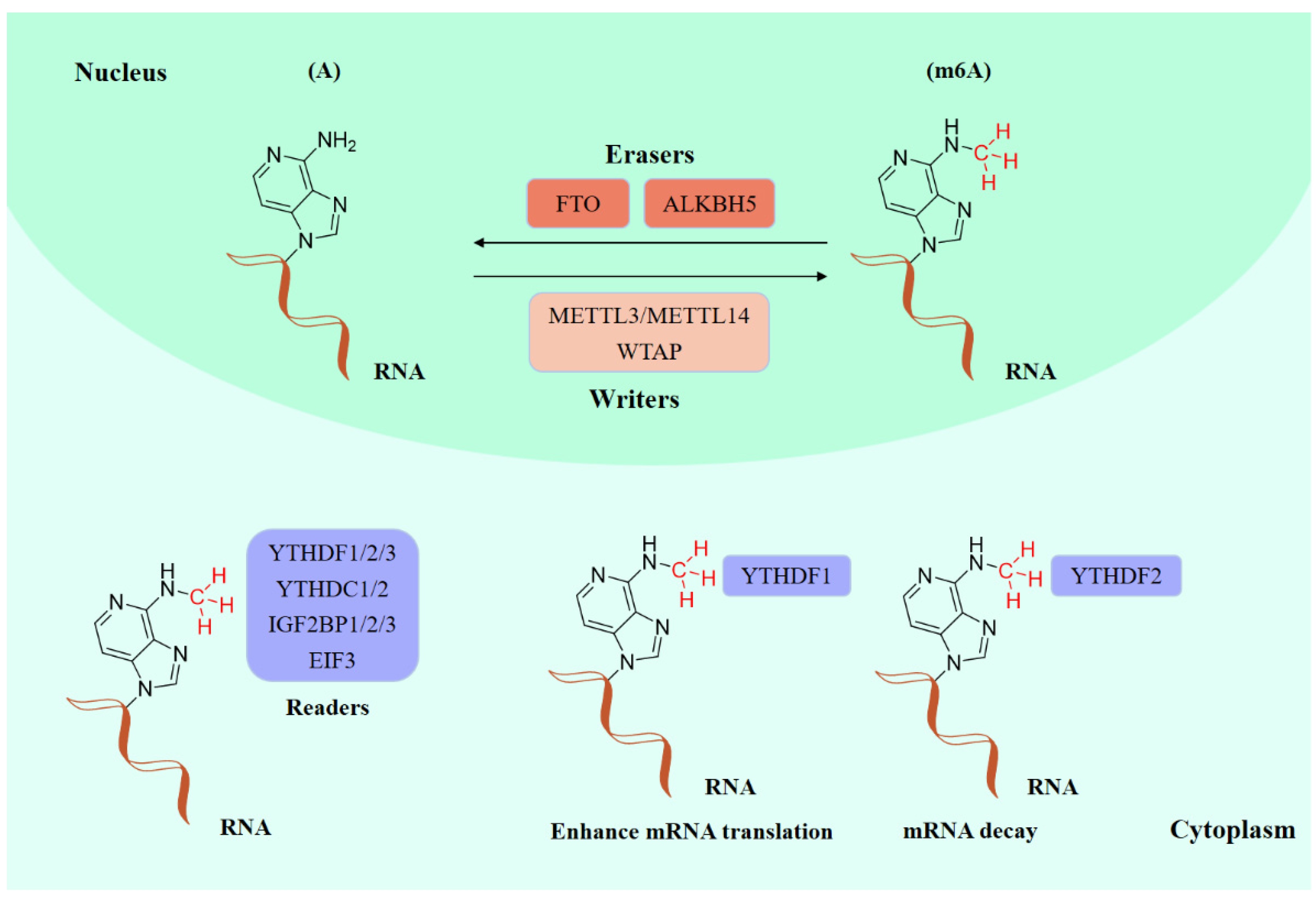
Figure 1. The molecular mechanism of m6A modifications.
In recent decades, epigenetic studies have explicitly highlighted the relationship between m6A demethylases and RNA metabolism, which can affect gene expression and animal development, as well as human disease progression [6][8][9]. FTO/ALKBH5 removes m6A modification with the assistance of cofactors 2-oxoglutarate (2OG) and Fe2+. They all belong to the AlkB subfamily of the 2OG dioxygenase superfamily [13][14]. Structurally, both FTO and ALKBH5 contain a highly conserved double-stranded β-helix (DSBH) fold (also called jelly-roll motif), providing a scaffold for the conserved ferrous ion (HXD/E…H motif) and 2OG binding site (Figure 2A–C). Moreover, structural insights into FTO and ALKBH5 identified some active-site residues that are critical to substrate-binding specificity and selectivity. These specific residues may help to design selective inhibitors against other AlkB family members.
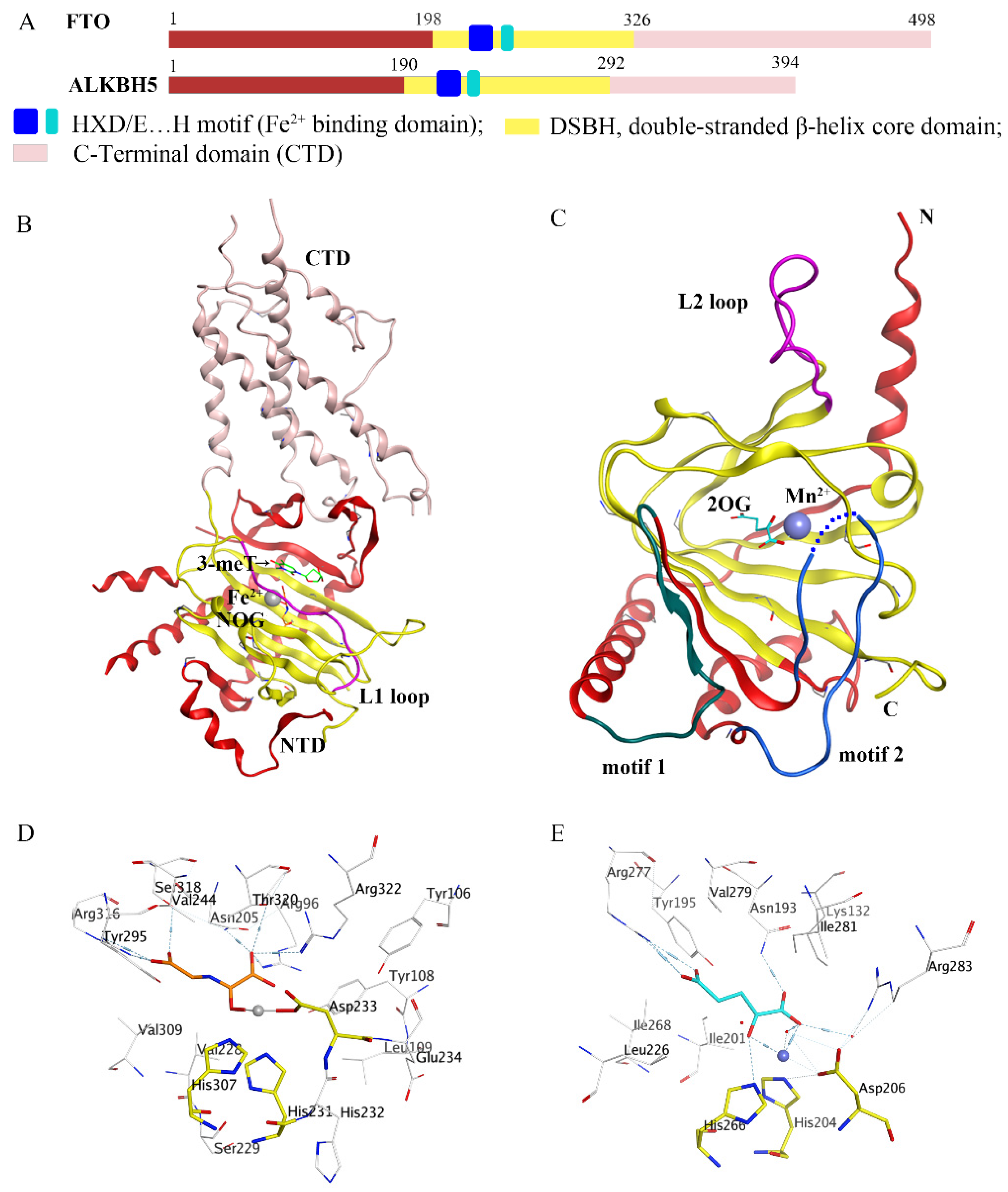
Figure 2. Comparison of fat-mass- and obesity-associated protein (FTO) and alkB homolog 5 (ALKBH5). (A) Structural domain representation of FTO and ALKBH5. (B) Crystal structure of FTOΔ31-3-meT (PDB ID: 3LFM) generated by MOE software. C-terminal domain (CTD) is colored in pink; N-terminal domain (NTD) is colored in red; the L1 loop is colored in purple; 3-meT is colored in green; N-oxalylglycine (NOG) is colored in orange; Fe2+ is colored in grey. (C) Crystal structure of ALKBH566-292-2OG (PDB ID: 4NRO) generated by MOE software; the motif 1 region is colored in dark green; the motif 2 region is colored in dark green; the L2 loop is colored in purple; 2-oxoglutarate (2OG) is colored in cyan, Mn2+ is colored in light blue; N: N-terminus, C: C-terminus. (D,E) The detailed interactions of the active center of FTO (PDB ID: 3LFM) and ALKBH5 (PDB ID: 4NRO) generated by MOE software. NOG and 2OG are colored in orange and cyan, respectively; Fe2+ and Mn2+ are drawn as grey and blue balls, respectively.
2. Inhibitors
2.1. Strategies Used for Developing m6A Demethylases Inhibitors
Since FTO and ALKBH5 rely on cofactors 2OG and Fe2+ for their m6A demethylation activity, early studies focused on screening a series of 2OG analogues and related compounds as their inhibitors [15]. Structure-based virtual screening of different compound libraries was an important way to obtain potent FTO/ALKBH5 inhibitors [16][17][18][19][20][21]. Interestingly, a high-throughput fluorescence polarization (FP) assay was performed for compounds that competed with FTO/ALKBH5 for binding to m6A-containing single-stranded nucleic acids, and meclofenamic acid (MA) was found to be a selective inhibitor of FTO over ALKBH5 [22]. Later on, Svensen and Jaffrey reported an approach to identify FTO inhibitors by using a fluorometric RNA substrate based on broccoli aptamer [23]. Das and co-workers designed a multi-protein dynamic combinatorial chemistry (DCC) system for screening FTO inhibitors [24]. More recently, Zhang et al. developed a single quantum dot-based Förster resonance energy transfer (FRET) nanosensor for FTO inhibitor screening [25]. Chang’s team identified several types of compounds that inhibit FTO activity through fluorescence quenching and molecular modeling studies [26][27][28]. Moreover, combining the information from crystal structures of ligand–protein complexes and structure-based drug designs was also an efficient approach to discover potent inhibitors with distinct chemical scaffolds [29][30][31].
2.2. FTO Inhibitors
2.2.1. Metal-Chelating Inhibitors
Since the 2OG dependent oxygenases shared the same protein-folding pattern, conserved cofactor, and substrate-binding sites, several 2OG oxygenases generic inhibitors have been shown to suppress FTO demethylation in vitro. These chemical inhibitors included 2OG, as well as pyridyl-, hydroxyquinoline-, and isoquinoline-based compounds [15][32]. Although they showed the different compound scaffolds, these inhibitors were all located in the 2OG binding pocket and chelated the metal ion in a bidentate manner. For instance, the 2OG competitive inhibitor 2-hydroxyglutarate (R-2HG, compound 1, Figure 3) has been identified as an FTO inhibitor, which displayed anticancer activity in leukemia and glioma [32][33]. N-oxalylglycine (NOG, compound 2, Figure 3), pyridine-2,4-dicarboxylate (2,4-PDCA, compound 3, Figure 3), and compound 4 (Figure 3) inhibited FTO demethylation in vitro, the IC50 values of which were 44, 8.3 and 15 μM, respectively [15].
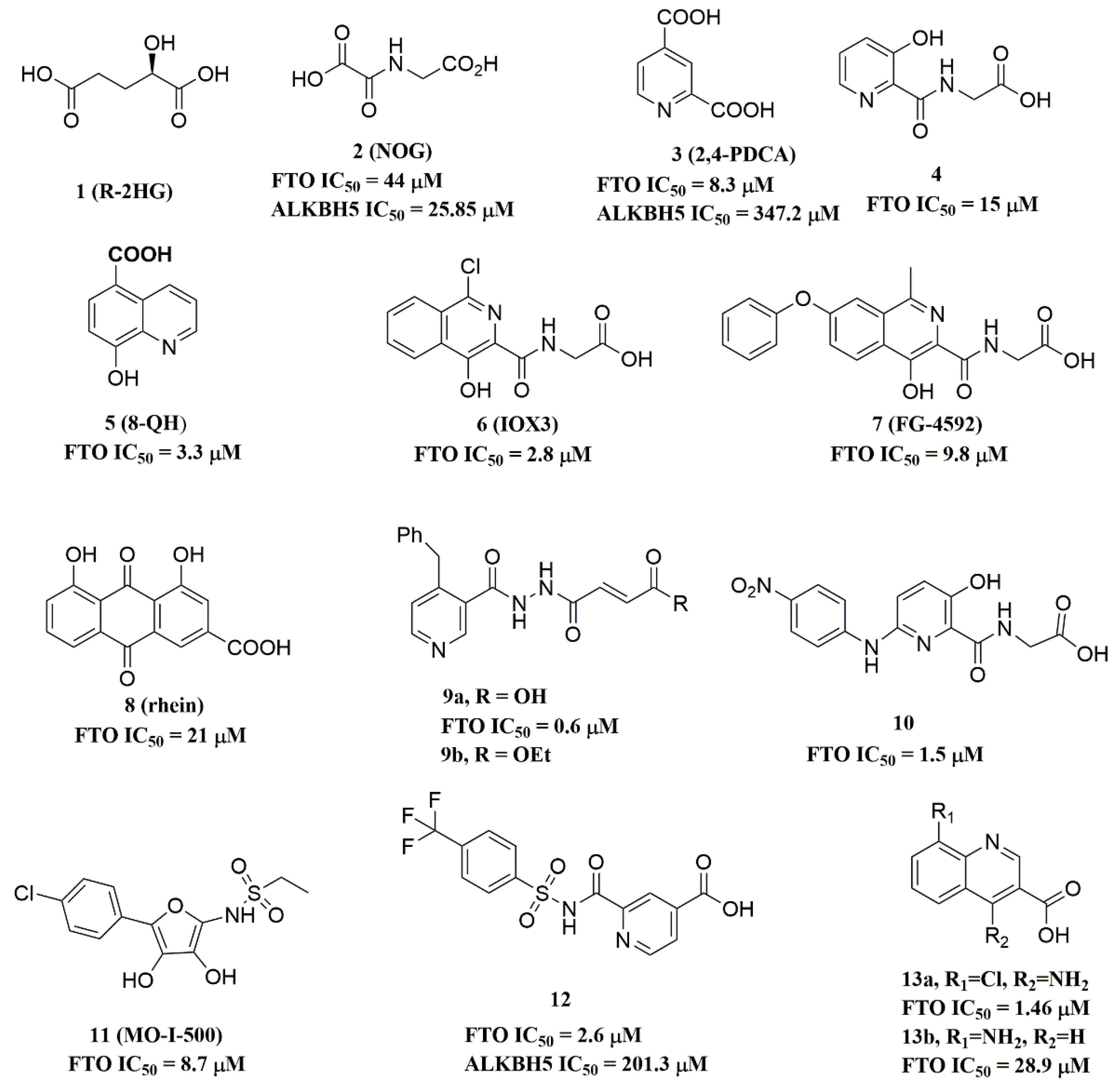
Figure 3. Metal-chelating inhibitors of FTO.
The 2D protein-ligand interaction diagrams were processed in four steps: (1) load the PDB file of the crystal complex into the MOE software; (2) rotate the crystal structure to a suitable angle and click “Compute” and “Ligand Interactions” buttons to create 2D diagrams; (3) change the “Legend” dropdown to “Rendering Options”, increase the residue size to 1.8 angstroms, and click “Apply”; and (4) save the diagram as an image in the TIF format with default parameters. Structures for FTO in complexes with NOG (PDB ID: 4IDZ) and 2,4-PDCA (PDB ID: 4IE0) showed that both of them are bound to metal ions (Figure 4A,B). Moreover, they further interacted with residues Arg316, Ser318, and Tyr295 of the side chains. In the complex of FTO with 4 (PDB ID: 4IE5), the pyridine ring of 4 nearly reached the substrate-binding site of FTO, which might spatially compete with the catalytic substrate (Figure 4C). Moreover, 8-QH (compound 5, Figure 3) was a relatively potent FTO inhibitor with an IC50 value of 3.3 μM. The crystal structure of FTO-8-QH (PDB ID: 4IE4) showed that 8-QH doubly chelated the Zn2+ ion with hydroxyl and nitrogen of the hydroxyquinoline in a similar way to NOG (Figure 4D). IOX3 (compound 6, Figure 3) and FG-4592 (compound 7, Figure 3) were known as prolyl-hydroxylase inhibitors [34], which also showed good inhibitory activity against FTO with IC50 of 2.8 and 9.8 μM, respectively [15][35]. The crystal structure of FTO-IOX3 (PDB ID: 4IE6) indicated that its chlorine atom of the isoquinoline group reached the substrate-binding site (Figure 4E).
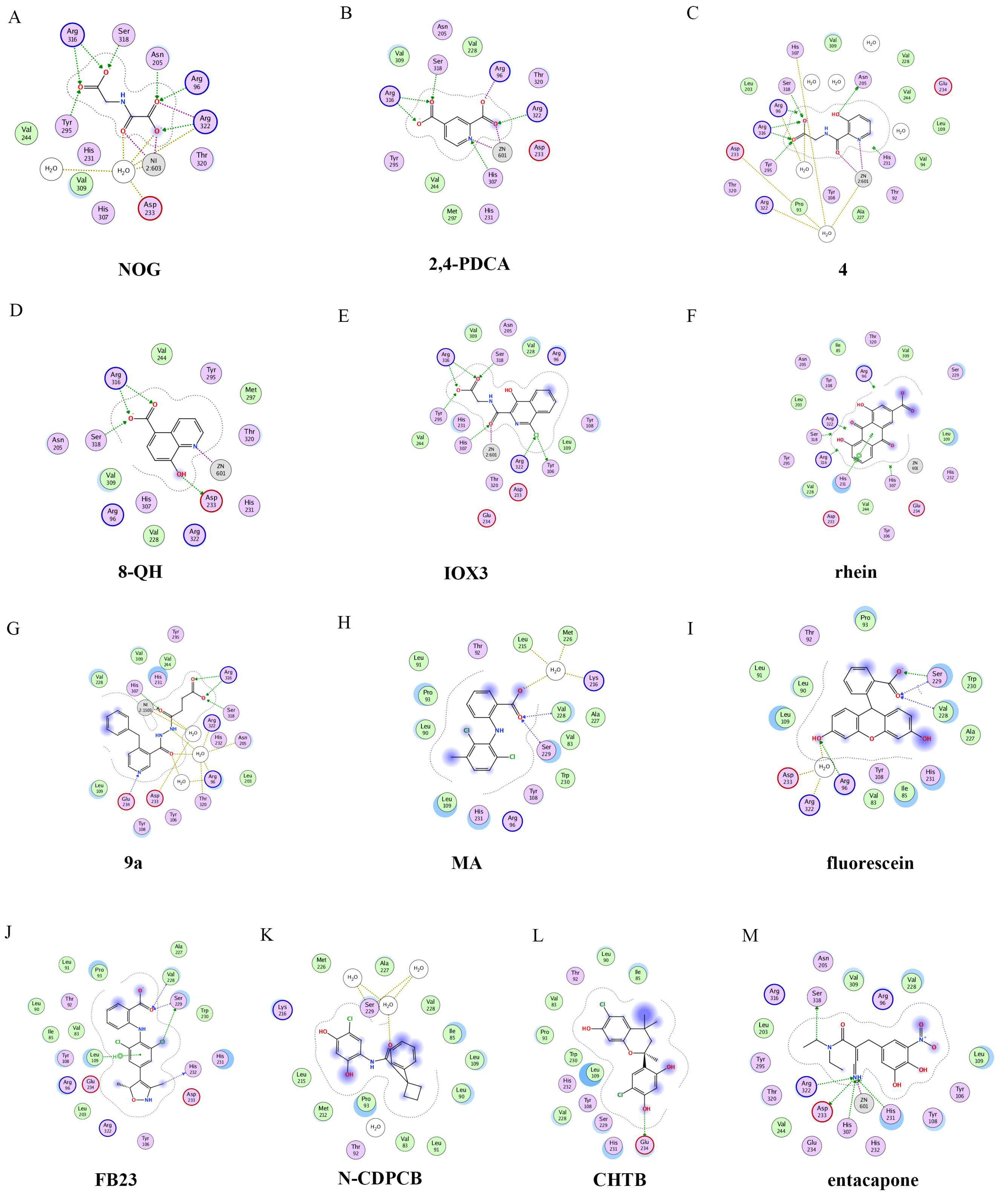
Figure 4. Two-dimensional representation of ligand–protein interactions of FTO inhibitors with FTO protein using MOE software. In MOE, the polar and non-polar residues are shown in pink and green disks; the water molecules are drawn as white circles; the metal ions are shown in grey circles; the hydrogen bonds are indicated by green dotted lines. (A–M) Ligand interactions of FTO with distinct compounds: (A) NOG (PDB ID: 4IDZ); (B) pyridine-2,4-dicarboxylate (2,4-PDCA, PDB ID: 4IE0); (C) 4 (PDB ID: 4IE5); (D) 8-QH (PDB ID: 4IE4); (E) IOX3 (PDB ID: 4IE6); (F) rhein (PDB ID: 4IE7); (G) 9a (PDB ID: 4CXW); (H) Meclofenamic acid (MA) (PDB ID: 4QKN); (I) fluorescein (PDB ID: 4ZS2); (J) FB23 (PDB ID: 6AKW); (K) N-CDPCB (PDB ID: 5DAB); (L) CHTB (PDB ID: 5F8P); and (M) entacapone (PDB ID: 6AK4).
In 2012, Chen et al. identified the natural product rhein (compound 8, Figure 3) (IC50 = 21 μM) as a competitive substrate inhibitor of FTO [17]. Further, rhein was the first discovered cell-active FTO inhibitor, which could inhibit cellular FTO demethylase activity. In molecular modeling of FTO-rhein (PDB ID: 4IE7), rhein occupied the binding sites of 3-meT, 2OG, and Fe2+. It is important to mention that this special structure blocked the binding of m6A containing ssDNA/ssRNA substrates to FTO (Figure 4F). Compound 9a (Figure 3) acted as a selective inhibitor of FTO (IC50 = 0.6 μM) compared to ALKBH5 (IC50 = 96.5 μM) and other AlkB subfamilies [36]. To view the superimposition from an FTO-3-meT-NOG (PDB ID: 3LFM) structure with that of FTO-9a (PDB ID: 4CXW), 9a occupied both 2OG and nucleotide acid binding sites (Figure 4G). The fumarate hydrazide of 9a was bound in the same combination as NOG, while the 4-benzyl pyridine side-chain sat in the nucleotide-binding site. Inferentially, the interaction between the pyridine nitrogen atom of 9a and Glu234 of FTO was the key factor for the high binding selectivity of FTO. In contrast, in other AlkB subfamilies, it was significantly weakened and even disappeared. In particular, both compound 9a and its ethyl ester derivative 9b (Figure 3) showed low cytotoxicity and significantly increased the global level of m6A in HeLa cells. Shishodia et al. used knowledge of the interaction of FTO with 2OG and substrates to design synthetic FTO inhibitors, of which compound 10 (IC50 = 1.5 μM, Figure 3) exhibited the best inhibitory activity [29].
Compound MO-I-500 (compound 11, Figure 3), a dihydroxyfuran sulfonamide [37], was the first identified as an FTO inhibitor, which displayed anticonvulsant activity. In the superposition of the MO-I-500 to NOG-FTO complex (PDB ID: 3LFM), this compound is located at the 2OG active site, and the hydroxyl oxygens of dihydroxyfuran chelated with the metal ion in opposite directions. The molecule MO-I-500 displayed anticonvulsant activity in the 6 Hz mouse model at a nontoxic dose, increased the total m6A level of cellular RNA, and altered the production of relative microRNAs. Through using a multi-protein DCC strategy, compound 12 (Figure 3) was identified as a FTO (IC50 = 2.6 μM) selective inhibitor, in comparison with ALKBH5 (IC50 = 201.3 μM) [24]. The structural model of FTO-12 revealed that compound 12 coordinated with Fe2+ in a bidentate manner, which was further stabilized by a combination of hydrogen-bonding and salt bridge interactions with Arg96, Arg319, Tyr295, and Ser318 of side chains from FTO. Two compounds 13a (IC50 = 1.46 µM, Figure 3) and 13b (IC50 = 28.9 µM, Figure 3) were defined as FTO inhibitors through a virtual screening on the ZINC compound library [18]. Molecular docking calculations revealed specific interactions between the amino acid residues of the FTO proteins Asp233, Tyr106, Glu234, Arg96, and Arg322, as well as two compounds. Importantly, compounds 13a and 13b are the first FTO inhibitors demonstrated to support the survival and rescue dopamine neurons from growth factor deprivation-induced apoptosis in vitro.
2.2.2. Substrate Competitive Inhibitors
Meclofenamic acid (MA) (compound 14a, Figure 5) and its derivatives were determined to be substrate competitive selective inhibitors of FTO [22][38][39]. When structural superimposition of the complexes of FTO-MA (PDB ID: 4QKN) and FTO-3-meT (PDB ID: 3LFM) was accomplished, in this case, MA partially covered the binding site of 3-meT in an L shape. In addition, there were stable hydrophobic interactions between a part of the FTO NRL and the carboxyl acid substituent of MA (Figure 4H) [22]. However, these hydrophobic interactions did not appear in the NRL of ALKBH5, which reduced the binding of MA to ALKBH5. MA2 (compound 14b, Figure 5), an ethyl ester derivative of MA, was a cell-active inhibitor of FTO, which could enhance the overall level of m6A in HeLa cells. Inspired by the specific binding of MA to FTO, fluorescein (compound 15a, Figure 5) and its derivatives, i.e., FL2 and FL2-DZ (compound 15b and compound 15c, Figure 5), were explored as FTO inhibitors with IC50 = 3.23 μM, 1.72 μM, and 4.49 μM, respectively. In FTO fluorescein’s (PDB ID: 4ZS2) crystal, fluorescein sat in the nucleotide-binding site of FTO, which was similar to MA (Figure 4I). Among them, FL2-DZ could selectively inhibit the demethylation of FTO. FL2-DZ also showed specific photo-affinity labeling of intracellular FTO because of the diazirine unit [39]. Thus, these fluorescein derivatives have dual functions of inhibiting FTO activity and labeling FTO. More recently, selective inhibitors FB23 (compound 16a, Figure 5) and FB23-2 (compound 16b, Figure 5) were synthesized by extending the dichloride-substituted benzene of MA. They were more efficient with IC50 values of 0.06 μM and 2.6 μM, respectively [38]. In the FTO-FB23 crystalline complex (PDB ID: 6AKW), FB23 occupied the entire binding position of MA in a similar L shape (Figure 4J). For FB23, the phenyl carboxylic acid substituent of MA was retained, forming several hydrophobic interactions with the nucleotide recognition cap. Hence, it showed the specific recognition capability of FTO compared to ALKBH5. Moreover, several hydrogen bonds were found between nitrogen or oxygen in the heterocyclic ring of FB23 and Glu234 of FTO, which was beneficial for the FB23 inhibitory activity of FTO. In vitro and in vivo research confirmed that FB23-2 improved the anti-proliferative activity of AML cell line cells and inhibited primary AML LSCs in mouse models.
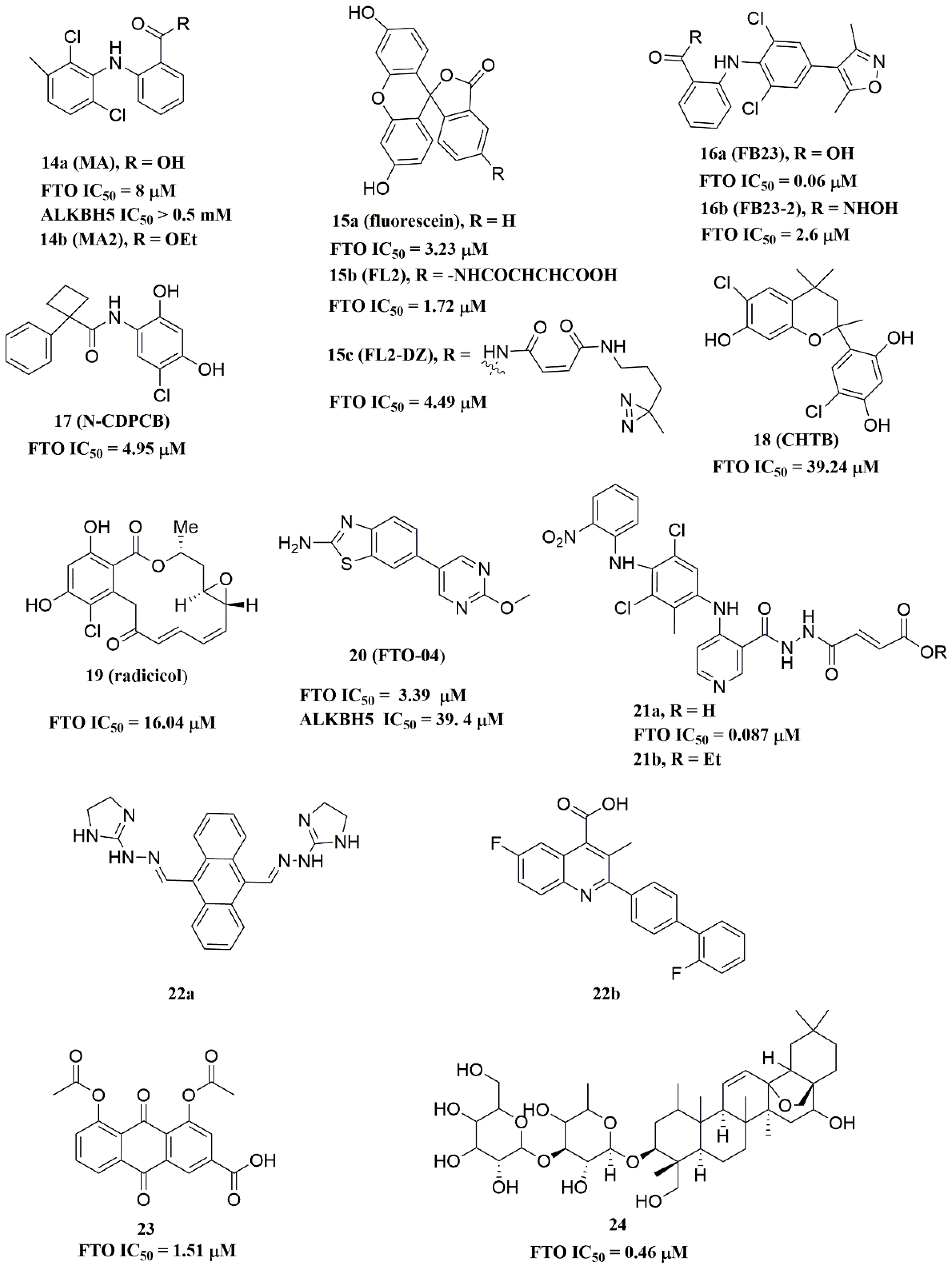
Figure 5. Substrate competitive inhibitors of FTO.
A series of benzene-1,3-diol derivatives were identified as selective inhibitors of FTO. They were N-CDPCB (compound 17, Figure 5) [30], CHTB (compound 18, Figure 5) [40], and radicicol (compound 19, Figure 5) [19]. IC50 values of N-CDPCB, CHTB and radicicol were 4.95 μM, 39.24 μM and 16.04 μM, respectively. In the crystal of compounds FTO-N-CDPCB (PDB ID: 5DAB) and N-CDPCB was sandwiched between the β-sheet and the L1 loop of FTO at the extension of the 2OG binding site (Figure 4K) [30]. In addition, the chlorine group was crucial for strengthening the N-CDPCB-FTO complex [30]. According to the binding pocket of N-CDPCB to FTO, a novel binding site was observed, which was partly overlapped with the inhibitor MA, not the 3-meT position. Interestingly, CHTB occupied the entire MA binding site in a similar L-shaped fashion in the crystal of FTO-CHTB (PDB ID: 5F8P) [40]. There were visible interactions between the chlorine atom in the chroman ring and several residues (Val83, Ile85, Leu90, and Thr92) of FTO in the hydrophobic pocket (Figure 4L). A hydrogen bond was also formed between residue Glu234 and the benzene hydroxyl group. Moreover, both N-CDPCB and CHTB were able to increase m6A abundance in total mRNA in 3T3-L1 cells. Inspired by the common structural features of N-CDPCB and CHTB, Chang’s group performed a structure-based virtual screening of compounds containing the 4-Cl-1,3-diol group and identified the natural compound radicicol as an effective FTO inhibitor [19]. Radicicol is bound to FTO and located at a similar cavity in the crystal of the FTO-radicicol complex, compared to N-CDPCB’s in an L-shaped conformation. One of the obvious differences between these two crystal complexes was that the conservative 4-Cl-1,3-diol group was bound to FTO in different orientations.
Additionally, by using Schrödinger software for molecular docking to target the MA binding site of FTO, a study designed and synthesized chemically distinct FTO inhibitors, of which FTO-04 (compound 20, Figure 5) was identified as a competitive inhibitor of FTO (IC50 = 3.39 μM) over ALKBH5 (IC50 = 39. 4 μM) [31]. Importantly, research demonstrated that FTO could impair the self-renewal properties of GSCs to inhibit neurosphere formation without altering the growth of human neural stem cell neurospheres. Prakash and co-workers synthesized compound 21a (Figure 5) as a potent FTO selectivity inhibitor (IC50 = 0.087 μM) by merging the key fragments of compound 9a and MA [41]. Moreover, the ester prodrug 21b of compound 21a could reduce the viability of AML cells by downregulating MYC and upregulating RARA, which was consistent with previous reports on the anticancer effect of pharmacological FTO inhibition [32][42].
In 2020, Chen and co-workers determined CS1 (compound 22a, Figure 5) and CS2 (compound 22b, Figure 5) as potent and selective FTO inhibitors through conducting a structure-based virtual screening [20]. Both CS1 and CS2 displayed a much higher anti-leukemic efficacy in comparison to FB23–2 in vitro and in vivo by modulating the expression of FTO target genes, including MYC, RARA, and ASB2. Moreover, this study also confirmed that CS1 and CS2 reprogramed immune response by reducing immune checkpoint gene expressions, especially leukocyte immunoglobulin-like receptor (LILRB4) [20]. In the same year, diacerein (compound 23, Figure 5) was identified as an FTO inhibitor by using a single quantum dot-based FRET nanosensor with an IC50 value of 1.51 μM [25]. Molecular modeling studies have suggested that diacerein possibly competed with m6A-containing ssDNA for FTO binding through forming hydrogen bonding with the amino acid residues of FTO protein. In addition, researchers validated the anti-proliferation effects of Saikosaponin-d (SsD, compound 24, Figure 5) in AML by targeting the m6A demethylation activity of FTO [43]. In vitro experiments showed that SsD exhibited good inhibitory activity on FTO demethylation with a low IC50 value of 0.46 μM. Importantly, they also demonstrated that SsD could overcome the resistance to tyrosine kinase inhibitors by suppressing FTO-mediated m6A RNA methylation pathways.
2.2.3. FTO Inhibitors with Other Scaffolds
Svensen and Jaffrey found a fluorogenic-methylated substrate for FTO based on the Broccoli aptamer [23]. After demethylation by FTO, this substrate was fluorescent. Subsequently, the fluorescent substrate was utilized to high-throughput screen FTO inhibitors with different chemical structures. As a consequence, a series of novel effective inhibitors were found, including amiloride analogue compound 25a (Figure 6), methionine derivative 25b (Figure 6), rhein analogues 25c-1 and 25c-2 (Figure 6), MA analogues 25d (Figure 6), and other scaffolds (25e-1, 25e-2, 25e-3, 25e-4, 25e-5) (Figure 6) with low IC50 values ranging from 0.34 μM to 3.00 μM. In comparison with ALKBH5, 25e-1 and 25e-4 showed selectivity for FTO. Further, 25c-2, 25e-1, and 25e-3 were cell-active in inhibiting FTO demethylase activity. These compounds provide new information for the design of more potent FTO inhibitors with new structural scaffolds.
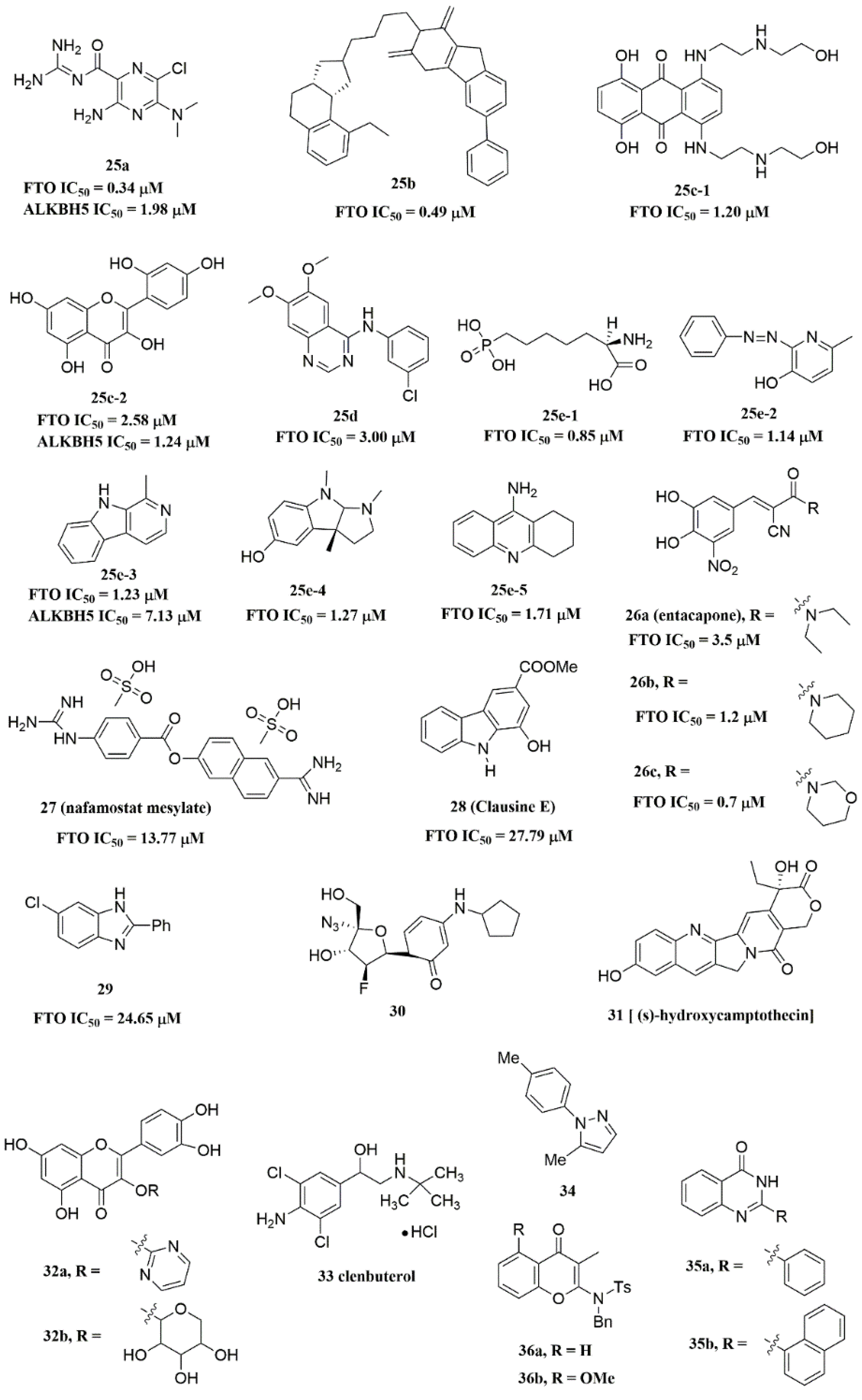
Figure 6. FTO inhibitors with different scaffolds.
Additionally, through structure-based virtual screening of U.S. Food and Drug Administration (FDA)-approved drugs, Peng et al. discovered that entacapone (compound 26a, Figure 6) was a substrate, as well as the 2OG cofactor competitive inhibitor of FTO [16]. Entacapone was structurally distinct from any reported inhibitors of FTO, the IC50 value of which was 3.5 μM. In the crystal of entacapone bound with FTO (PDB ID: 6AK4), hydrogen bonds could be discovered between the heterotopic hydroxyl group on the nitrocatechol ring with residues from the substrate-binding site (Figure 4M). Additionally, the nitrile group of the compound could chelate with Zn2+, which was recently reported in histone demethylase protein–ligand complex cases. Interestingly, the flexible tail of diethyl-propanamide was embedded deeply in the cofactor binding site. Furthermore, compounds 26b (Figure 6) and 26c (Figure 6) were designed and synthesized by replacing the flexible diethyl tail of entacapone with alicyclic groups, enhancing the inhibitory activity of FTO with IC50 values of 1.2 and 0.7 μM, respectively.
Combining the fluorescence quenching technology, several inhibitors were found to decrease the demethylase activity of FTO, including nafamostat mesylate (compound 27, Figure 6) [26], clausine E (compound 28, Figure 6) [27], 2-phenyl-1H-benzimidazole 29 (Figure 6) [28], fluoronucleoside analogue 30 (Figure 6) [44], (s)-hydroxycamptothecin (compound 31, Figure 6) [45], flavonols 32a and 32b (Figure 6) [46], clenbuterol 33 (Figure 6) [47], pyrazole derivative 34 (Figure 6) [48], 1,3-diazaheterocyclic compounds 35a and 35b (Figure 6) [49], and 3-substituted 2-aminochromones 36a and 36b (Figure 6) [50]. Among them, nafamostat mesylate, clausine E, and compound 29 showed good inhibitory activity against FTO with IC50 values of 13.77 μM, 27.79 μM, and 24.65 μM, respectively. Moreover, molecular docking model analysis showed that the affinity bindings between FTO and these molecules were mainly forced by the hydrophobic and hydrogen bonds interactions with residues from the active cavity of FTO, which were similar to the binding modes between FTO and other inhibitors.
2.3. ALKBH5 Inhibitors
Recently, several research groups found that 2OG inhibitors showed the inhibiting activity of ALKBH5 demethylase. In these cases, the crystalline complexes of ALKBH5 with different compounds were also obtained to display the direct structural evidence [51][52][53][54]. For instance, NOG and succinate (compound 37, Figure 7) showed IC50 values of 25.85 μM and 30.00 μM, respectively. However, different from FTO, 2,4-PDCA (IC50 = 347.2 μM) and citrate (compound 38, IC50 = 488 μM) were moderate inhibitors of ALKBH5 (Figure 7). In the ALKBH5 crystal with NOG, succinate, and 2,4-PDCA (PDB IDs: 4NRP [52], 4NPM [54], or 4NRQ [52]), all of the corresponding inhibitors were located in the 2OG active site of ALKBH5 and chelated with Mn2+ (Figure 8A–C). Moreover, according to the overlay of ALKBH5-citrate (PDB entry 4O61) and FTO-citrate (PDB entry 4IE7), the citrate molecule competed with 2OG, and the 2OG binding sites were partially covered by its positions (Figure 8D) [53]. One possible reason was that the residues Ile281 and Tyr195 of ALKBH5 blocked citrate from reaching the 2OG active site. Notably, even if citrate displayed the modest inhibitory activity on the ALKBH family dioxygenases, the discrepancies of citrate binding to FTO and ALKBH5 could provide a strategy to design new types of ALKBH5-specific inhibitors.
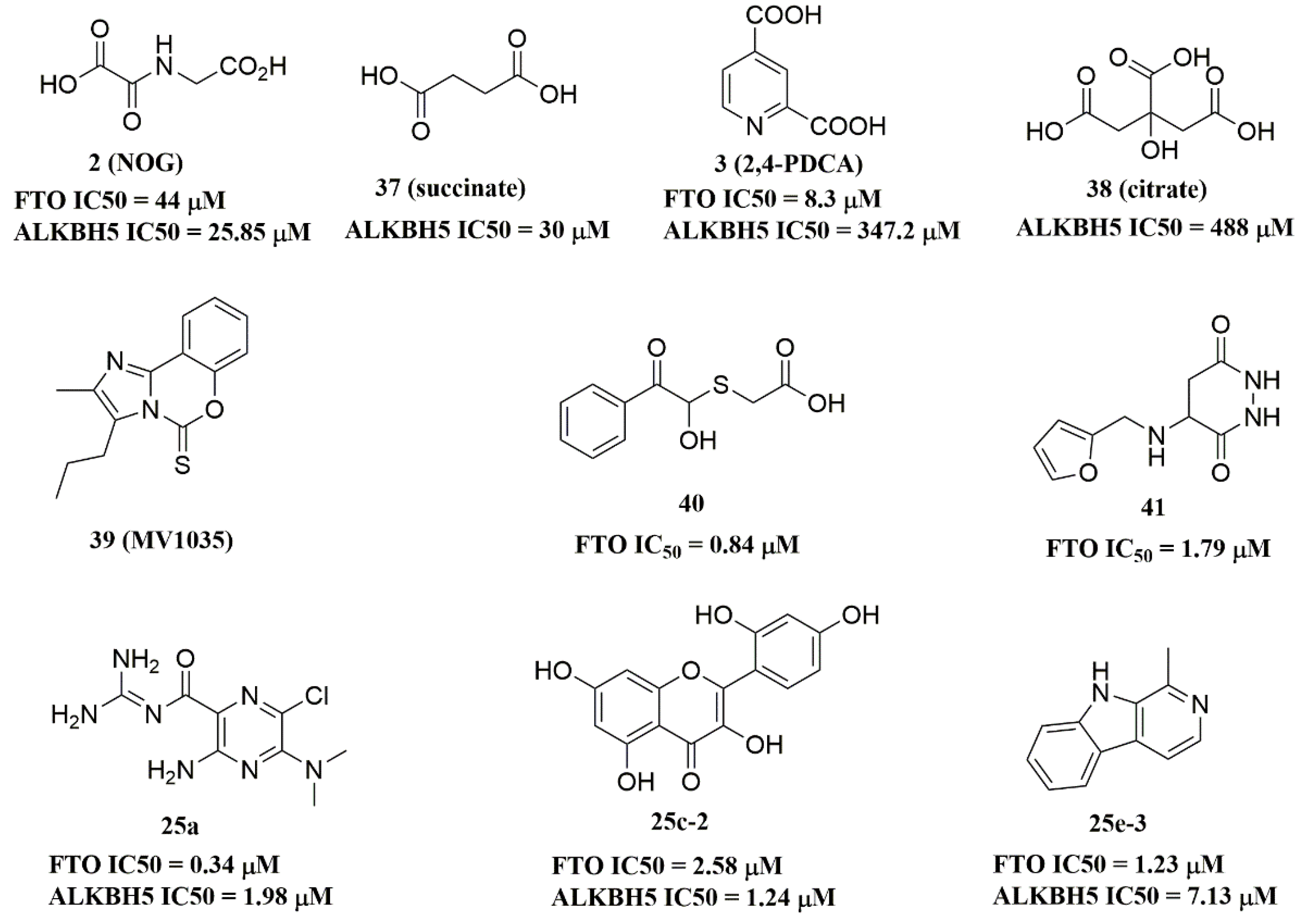
Figure 7. ALKBH5 inhibitors.
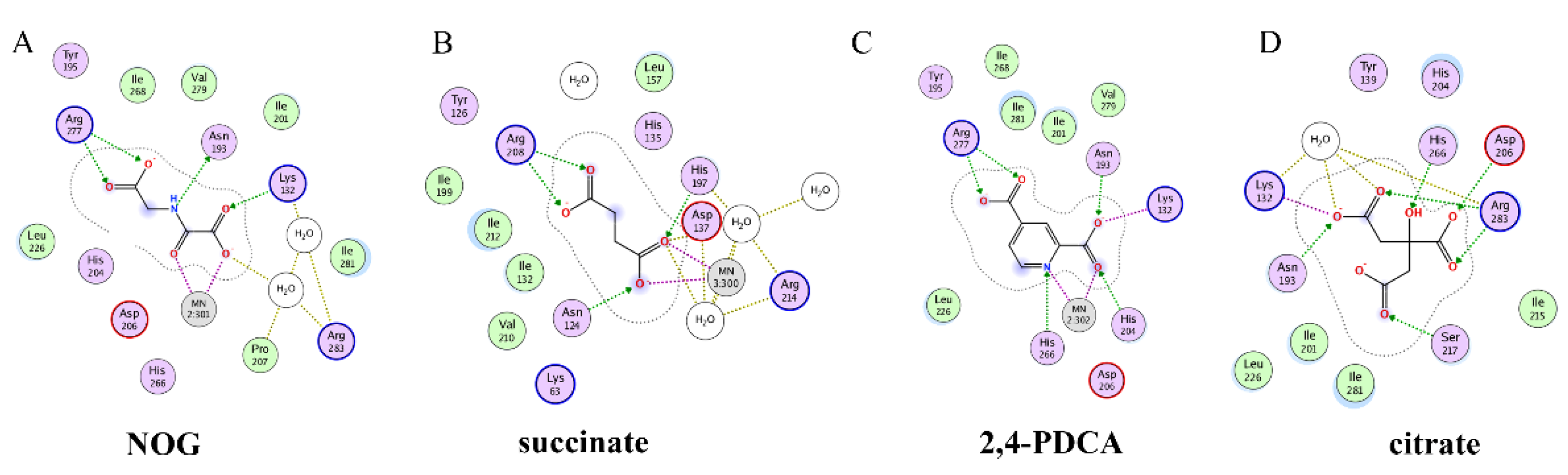
Figure 8. Two-dimensional representation of ligand–protein interactions of ALKBH5 inhibitors with ALKBH5 protein using MOE software. (A–D) Crystal structures of ALKBH5 with different compounds: (A) NOG (PDB ID: 4NRP); (B) succinate (PDB ID: 4NPM); (C) 2,4-PDCA (PDB ID: 4NRQ); (D) citrate (PDB ID: 4O61).
The imidazobenzoxazin-5-thione MV1035 (compound 39, Figure 7) was demonstrated to inhibit ALKBH5 demethylase activity in vitro. At the same time, it significantly reduced the migration and invasiveness of the U87 glioblastoma cell lines [55]. In this case, they reported a potential binding site for MV1035 within ALKBH5. MV1035 overlapped with the catalytic site of the enzyme, specifically near the carboxylate group of 2OG. Recently, a structure-based virtual screening identified two compounds, 2-[(1-hydroxy-2-oxo-2-phenylethyl)sulfanyl]acetic acid(compound 40, Figure 7)) and 4-{[(furan-2-yl)methyl]amino}-1,2-diazinane-3,6-dione (compound 41, Figure 7)), which acted as ALKBH5 inhibitors with IC50 values of 0.84 μM and 1.79 μM, respectively [21]. This study also demonstrated that these two inhibitors suppressed the cell proliferation of several AML cell lines at low micromolar concentrations, with IC50 ranging from 1.38 to 16.5 μM. In addition, some FTO inhibitors also showed similar activity with ALKBH5. For example, amiloride analogue 25a (Figure 7), rhein analog 25c-2 (Figure 7), and compound 25e-3 (Figure 7) showed inhibitory activity with IC50 values of 1.98 μM, 7.13 μM, and 1.24 μM for target ALKBH5, respectively.
References
- Boccaletto, P.; Machnicka, M.A.; Purta, E.; Piatkowski, P.; Baginski, B.; Wirecki, T.K.; de Crécy-Lagard, V.; Ross, R.; Limbach, P.A.; Kotter, A.; et al. MODOMICS: A database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018, 46, D303–D307.
- Zhao, B.S.; Roundtree, I.A.; He, C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 2017, 18, 31–42.
- Niu, Y.; Zhao, X.; Wu, Y.S.; Li, M.M.; Wang, X.J.; Yang, Y.G. N6-methyl-adenosine (m6A) in RNA: An old modification with a novel epigenetic function. Genom. Proteom. Bioinform. 2013, 11, 8–17.
- Desrosiers, R.; Friderici, K.; Rottman, F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc. Natl. Acad. Sci. USA 1974, 71, 3971–3975.
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62.
- Shi, H.; Wei, J.; He, C. Where, When, and How: Context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers. Mol. Cell 2019, 74, 640–650.
- Zaccara, S.; Ries, R.J.; Jaffrey, S.R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 608–624.
- Ma, S.; Chen, C.; Ji, X.; Liu, J.; Zhou, Q.; Wang, G.; Yuan, W.; Kan, Q.; Sun, Z. The interplay between m6A RNA methylation and noncoding RNA in cancer. J. Hematol. Oncol. 2019, 12, 121.
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.G.; et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887.
- Zheng, G.; Dahl, J.A.; Niu, Y.; Fedorcsak, P.; Huang, C.M.; Li, C.J.; Vågbø, C.B.; Shi, Y.; Wang, W.L.; Song, S.H.; et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 2013, 49, 18–29.
- He, L.; Li, H.; Wu, A.; Peng, Y.; Shu, G.; Yin, G. Functions of N6-methyladenosine and its role in cancer. Mol. Cancer 2019, 18, 176.
- Yang, Y.; Hsu, P.J.; Chen, Y.S.; Yang, Y.G. Dynamic transcriptomic m(6)A decoration: Writers, erasers, readers and functions in RNA metabolism. Cell Res. 2018, 28, 616–624.
- Gerken, T.; Girard, C.A.; Tung, Y.C.; Webby, C.J.; Saudek, V.; Hewitson, K.S.; Yeo, G.S.; McDonough, M.A.; Cunliffe, S.; McNeill, L.A.; et al. The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science 2007, 318, 1469–1472.
- Thalhammer, A.; Bencokova, Z.; Poole, R.; Loenarz, C.; Adam, J.; O’Flaherty, L.; Schödel, J.; Mole, D.; Giaslakiotis, K.; Schofield, C.J.; et al. Human AlkB homologue 5 is a nuclear 2-oxoglutarate dependent oxygenase and a direct target of hypoxia-inducible factor 1α (HIF-1α). PLoS ONE 2011, 6, e16210.
- Aik, W.; Demetriades, M.; Hamdan, M.K.; Bagg, E.A.; Yeoh, K.K.; Lejeune, C.; Zhang, Z.; McDonough, M.A.; Schofield, C.J. Structural basis for inhibition of the fat mass and obesity associated protein (FTO). J. Med. Chem. 2013, 56, 3680–3688.
- Peng, S.; Xiao, W.; Ju, D.; Sun, B.; Hou, N.; Liu, Q.; Wang, Y.; Zhao, H.; Gao, C.; Zhang, S.; et al. Identification of entacapone as a chemical inhibitor of FTO mediating metabolic regulation through FOXO1. Sci. Transl. Med. 2019, 11, eaau7116.
- Chen, B.; Ye, F.; Yu, L.; Jia, G.; Huang, X.; Zhang, X.; Peng, S.; Chen, K.; Wang, M.; Gong, S.; et al. Development of cell-active N6-methyladenosine RNA demethylase FTO inhibitor. J. Am. Chem. Soc. 2012, 134, 17963–17971.
- Selberg, S.; Yu, L.Y.; Bondarenko, O.; Kankuri, E.; Seli, N.; Kovaleva, V.; Herodes, K.; Saarma, M.; Karelson, M. Small-Molecule Inhibitors of the RNA M6A Demethylases FTO Potently Support the Survival of Dopamine Neurons. Int. J. Mol. Sci. 2021, 22, 4537.
- Wang, R.; Han, Z.; Liu, B.; Zhou, B.; Wang, N.; Jiang, Q.; Qiao, Y.; Song, C.; Chai, J.; Chang, J. Identification of Natural Compound Radicicol as a Potent FTO Inhibitor. Mol. Pharm. 2018, 15, 4092–4098.
- Su, R.; Dong, L.; Li, Y.; Gao, M.; Han, L.; Wunderlich, M.; Deng, X.; Li, H.; Huang, Y.; Gao, L.; et al. Targeting FTO Suppresses Cancer Stem Cell Maintenance and Immune Evasion. Cancer Cell 2020, 38, 79–96.e11.
- Selberg, S.; Seli, N.; Kankuri, E.; Karelson, M. Rational Design of Novel Anticancer Small-Molecule RNA m6A Demethylase ALKBH5 Inhibitors. ACS Omega 2021, 6, 13310–13320.
- Huang, Y.; Yan, J.; Li, Q.; Li, J.; Gong, S.; Zhou, H.; Gan, J.; Jiang, H.; Jia, G.F.; Luo, C.; et al. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. 2015, 43, 373–384.
- Svensen, N.; Jaffrey, S.R. Fluorescent RNA Aptamers as a Tool to Study RNA-Modifying Enzymes. Cell Chem. Biol. 2016, 23, 415–425.
- Das, M.; Yang, T.; Dong, J.; Prasetya, F.; Xie, Y.; Wong, K.H.Q.; Cheong, A.; Woon, E.C.Y. Multiprotein Dynamic Combinatorial Chemistry: A Strategy for the Simultaneous Discovery of Subfamily-Selective Inhibitors for Nucleic Acid Demethylases FTO and ALKBH3. Chem. Asian J. 2018, 13, 2854–2867.
- Zhang, Y.; Li, Q.N.; Zhou, K.; Xu, Q.; Zhang, C.Y. Identification of Specific N(6)-Methyladenosine RNA Demethylase FTO Inhibitors by Single-Quantum-Dot-Based FRET Nanosensors. Anal. Chem. 2020, 92, 13936–13944.
- Han, X.; Wang, N.; Li, J.; Wang, Y.; Wang, R.; Chang, J. Identification of nafamostat mesilate as an inhibitor of the fat mass and obesity-associated protein (FTO) demethylase activity. Chem. Biol. Interact. 2019, 297, 80–84.
- Wang, Y.; Li, J.; Han, X.; Wang, N.; Song, C.; Wang, R.; Chang, J. Identification of Clausine E as an inhibitor of fat mass and obesity-associated protein (FTO) demethylase activity. J. Mol. Recognit. 2019, 32, e2800.
- Li, J.; Wang, Y.; Han, X.; Wang, N.; Yu, W.; Wang, R.; Chang, J. The role of chlorine atom on the binding between 2-phenyl-1H-benzimidazole analogues and fat mass and obesity-associated protein. J. Mol. Recognit. 2019, 32, e2774.
- Shishodia, S.; Demetriades, M.; Zhang, D.; Tam, N.Y.; Maheswaran, P.; Clunie-O’Connor, C.; Tumber, A.; Leung, I.K.H.; Ng, Y.M.; Leissing, T.M.; et al. Structure-Based Design of Selective Fat Mass and Obesity Associated Protein (FTO) Inhibitors. J. Med. Chem. 2021, 64, 16609–16625.
- He, W.; Zhou, B.; Liu, W.; Zhang, M.; Shen, Z.; Han, Z.; Jiang, Q.; Yang, Q.; Song, C.; Wang, R.; et al. Identification of A Novel Small-Molecule Binding Site of the Fat Mass and Obesity Associated Protein (FTO). J. Med. Chem. 2015, 58, 7341–7348.
- Huff, S.; Tiwari, S.K.; Gonzalez, G.M.; Wang, Y.; Rana, T.M. m(6)A-RNA Demethylase FTO Inhibitors Impair Self-Renewal in Glioblastoma Stem Cells. ACS Chem. Biol. 2021, 16, 324–333.
- Su, R.; Dong, L.; Li, C.; Nachtergaele, S.; Wunderlich, M.; Qing, Y.; Deng, X.; Wang, Y.; Weng, X.; Hu, C.; et al. R-2HG Exhibits Anti-tumor Activity by Targeting FTO/m(6)A/MYC/CEBPA Signaling. Cell 2018, 172, 90–105.e23.
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30.
- Rose, N.R.; McDonough, M.A.; King, O.N.; Kawamura, A.; Schofield, C.J. Inhibition of 2-oxoglutarate dependent oxygenases. Chem Soc. Rev. 2011, 40, 4364–4397.
- McMurray, F.; Demetriades, M.; Aik, W.; Merkestein, M.; Kramer, H.; Andrew, D.S.; Scudamore, C.L.; Hough, T.A.; Wells, S.; Ashcroft, F.M.; et al. Pharmacological inhibition of FTO. PLoS ONE 2015, 10, e0121829.
- Toh, J.D.W.; Sun, L.; Lau, L.Z.M.; Tan, J.; Low, J.J.A.; Tang, C.W.Q.; Cheong, E.J.Y.; Tan, M.J.H.; Chen, Y.; Hong, W.; et al. A strategy based on nucleotide specificity leads to a subfamily-selective and cell-active inhibitor of N(6)-methyladenosine demethylase FTO. Chem. Sci. 2015, 6, 112–122.
- Zheng, G.; Cox, T.; Tribbey, L.; Wang, G.Z.; Iacoban, P.; Booher, M.E.; Gabriel, G.J.; Zhou, L.; Bae, N.; Rowles, J.; et al. Synthesis of a FTO inhibitor with anticonvulsant activity. ACS Chem. Neurosci. 2014, 5, 658–665.
- Huang, Y.; Su, R.; Sheng, Y.; Dong, L.; Dong, Z.; Xu, H.; Ni, T.; Zhang, Z.S.; Zhang, T.; Li, C.; et al. Small-Molecule Targeting of Oncogenic FTO Demethylase in Acute Myeloid Leukemia. Cancer Cell 2019, 35, 677–691.e10.
- Wang, T.; Hong, T.; Huang, Y.; Su, H.; Wu, F.; Chen, Y.; Wei, L.; Huang, W.; Hua, X.; Xia, Y.; et al. Fluorescein Derivatives as Bifunctional Molecules for the Simultaneous Inhibiting and Labeling of FTO Protein. J. Am. Chem. Soc. 2015, 137, 13736–13739.
- Qiao, Y.; Zhou, B.; Zhang, M.; Liu, W.; Han, Z.; Song, C.; Yu, W.; Yang, Q.; Wang, R.; Wang, S.; et al. A Novel Inhibitor of the Obesity-Related Protein FTO. Biochemistry 2016, 55, 1516–1522.
- Prakash, M.; Itoh, Y.; Fujiwara, Y.; Takahashi, Y.; Takada, Y.; Mellini, P.; Elboray, E.E.; Terao, M.; Yamashita, Y.; Yamamoto, C.; et al. Identification of Potent and Selective Inhibitors of Fat Mass Obesity-Associated Protein Using a Fragment-Merging Approach. J. Med. Chem. 2021, 64, 15810–15824.
- Li, Z.; Weng, H.; Su, R.; Weng, X.; Zuo, Z.; Li, C.; Huang, H.; Nachtergaele, S.; Dong, L.; Hu, C.; et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N(6)-Methyladenosine RNA Demethylase. Cancer Cell 2017, 31, 127–141.
- Sun, K.; Du, Y.; Hou, Y.; Zhao, M.; Li, J.; Du, Y.; Zhang, L.; Chen, C.; Yang, H.; Yan, F.; et al. Saikosaponin D exhibits anti-leukemic activity by targeting FTO/m(6)A signaling. Theranostics 2021, 11, 5831–5846.
- Li, Z.; Wang, Z.; Wang, N.; Han, X.; Yu, W.; Wang, R.; Chang, J. Identification of the binding between three fluoronucleoside analogues and fat mass and obesity-associated protein by isothermal titration calorimetry and spectroscopic techniques. J. Pharm. Biomed. Anal. 2018, 149, 290–295.
- Ren, T.; Wang, Z.; Zhang, L.; Wang, N.; Han, X.; Wang, R.; Chang, J. Study of the Binding between Camptothecin Analogs and FTO by Spectroscopy and Molecular Docking. J. Fluoresc. 2017, 27, 1467–1477.
- Wang, Z.; Wang, N.; Han, X.; Wang, R.; Chang, J. Interaction of two flavonols with fat mass and obesity-associated protein investigated by fluorescence quenching and molecular docking. J. Biomol. Struct. Dyn. 2018, 36, 3388–3397.
- Wang, Z.; Han, X.; Wang, N.; Wang, R.; Chang, J. Binding of clenbuterol to HSA and FTO: A spectroscopic analysis and molecular docking. Med. Chem. Res. 2018, 27, 944–953.
- Yang, L.; Yu, W.; Li, Z.; Zhang, X.; Ren, T.; Zhang, L.; Wang, R.; Chang, J. Comparative study of the binding between FTO protein and five pyrazole derivatives by spectrofluorimetry. J. Mol. Liq. 2016, 218, 156–165.
- Zhang, L.; Ren, T.; Tian, X.; Wang, Z.; Yu, W.; Wang, R.; Chang, J. Investigation of the Interaction between 1,3-Diazaheterocyclic Compounds and the Fat Mass and Obesity-Associated Protein by Fluorescence Spectroscopy and Molecular Modeling. J. Fluoresc. 2017, 27, 369–378.
- Zhang, L.; Wang, Z.; Ren, T.; Liu, H.; Wang, X.; Wang, R.; Chang, J. Investigation of the interaction between FTO and 3-substituted 2-aminochromones by spectroscopy and molecular modeling. Med. Chem. Res. 2017, 26, 1349–1358.
- Aik, W.; Scotti, J.S.; Choi, H.; Gong, L.; Demetriades, M.; Schofield, C.J.; McDonough, M.A. Structure of human RNA N⁶-methyladenine demethylase ALKBH5 provides insights into its mechanisms of nucleic acid recognition and demethylation. Nucleic Acids Res. 2014, 42, 4741–4754.
- Feng, C.; Liu, Y.; Wang, G.; Deng, Z.; Zhang, Q.; Wu, W.; Tong, Y.; Cheng, C.; Chen, Z. Crystal structures of the human RNA demethylase Alkbh5 reveal basis for substrate recognition. J. Biol. Chem. 2014, 289, 11571–11583.
- Xu, C.; Liu, K.; Tempel, W.; Demetriades, M.; Aik, W.; Schofield, C.J.; Min, J. Structures of human ALKBH5 demethylase reveal a unique binding mode for specific single-stranded N6-methyladenosine RNA demethylation. J. Biol. Chem. 2014, 289, 17299–17311.
- Chen, W.; Zhang, L.; Zheng, G.; Fu, Y.; Ji, Q.; Liu, F.; Chen, H.; He, C. Crystal structure of the RNA demethylase ALKBH5 from zebrafish. FEBS Lett. 2014, 588, 892–898.
- Malacrida, A.; Rivara, M.; Di Domizio, A.; Cislaghi, G.; Miloso, M.; Zuliani, V.; Nicolini, G. 3D proteome-wide scale screening and activity evaluation of a new ALKBH5 inhibitor in U87 glioblastoma cell line. Bioorg. Med. Chem. 2020, 28, 115300.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.3K
Revisions:
2 times
(View History)
Update Date:
06 Jun 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No