 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | William Blalock | -- | 3954 | 2022-06-03 14:01:56 | | | |
| 2 | Beatrix Zheng | -33 word(s) | 3921 | 2022-06-06 10:53:22 | | |
Video Upload Options
Since first being documented in ancient times, the relation of inflammation with injury and disease has evolved in complexity and causality. Early observations supported a cause (injury) and effect (inflammation) relationship, but the number of pathologies linked to chronic inflammation suggests that inflammation itself acts as a potent promoter of injury and disease. Additionally, results from studies over the last decades point to chronic inflammation and innate immune signaling as a critical link between stress (exogenous and endogenous) and adaptation.
1. Overview
Since first being documented in ancient times by Greek and Egyptian physicians, the relationship of inflammation with injury and disease has evolved in complexity as well as causality. Initial characteristic descriptions by Hippocrates (5th century BC) included swelling or “edema” of the affected tissue. Later, Aulus Celsus (1st century BC/1st century AD) described the four main manifestations of inflammation: pain, edema, warmth, and redness of the interested tissue. The Roman physician/surgeon Galen later added the fifth hallmark of inflammation—loss of function of the affected tissue—a hallmark most closely associated with muscular, bone, or joint manifestations of inflammation visible, at the time, upon examination of the patient [1]. The empirical observations set the framework for detailed scientific studies over the centuries that resulted in the discovery and characterization of the immune system and its role in the response to not only injury, but also disease. These early studies supported a cause-and-effect theory whereby injury or disease gave rise to inflammation. However, is inflammation simply a case of the immune system responding to disease or can inflammation have a causative role in disease? The number of pathologies now linked to chronic inflammation would suggest inflammation may be able to both promote and be the initiating stimulus in multiple pathologies, but to this day the inflammation–disease relationship remains controversial. To clearly understand this relationship, one must first understand the relationship between inflammation, innate and acquired (or cell-based) immunity, DNA repair, and the assortment of mutations and epigenetic modifications accumulated during chronic inflammation.
2. Innate Immunity vs. Adaptive Immunity
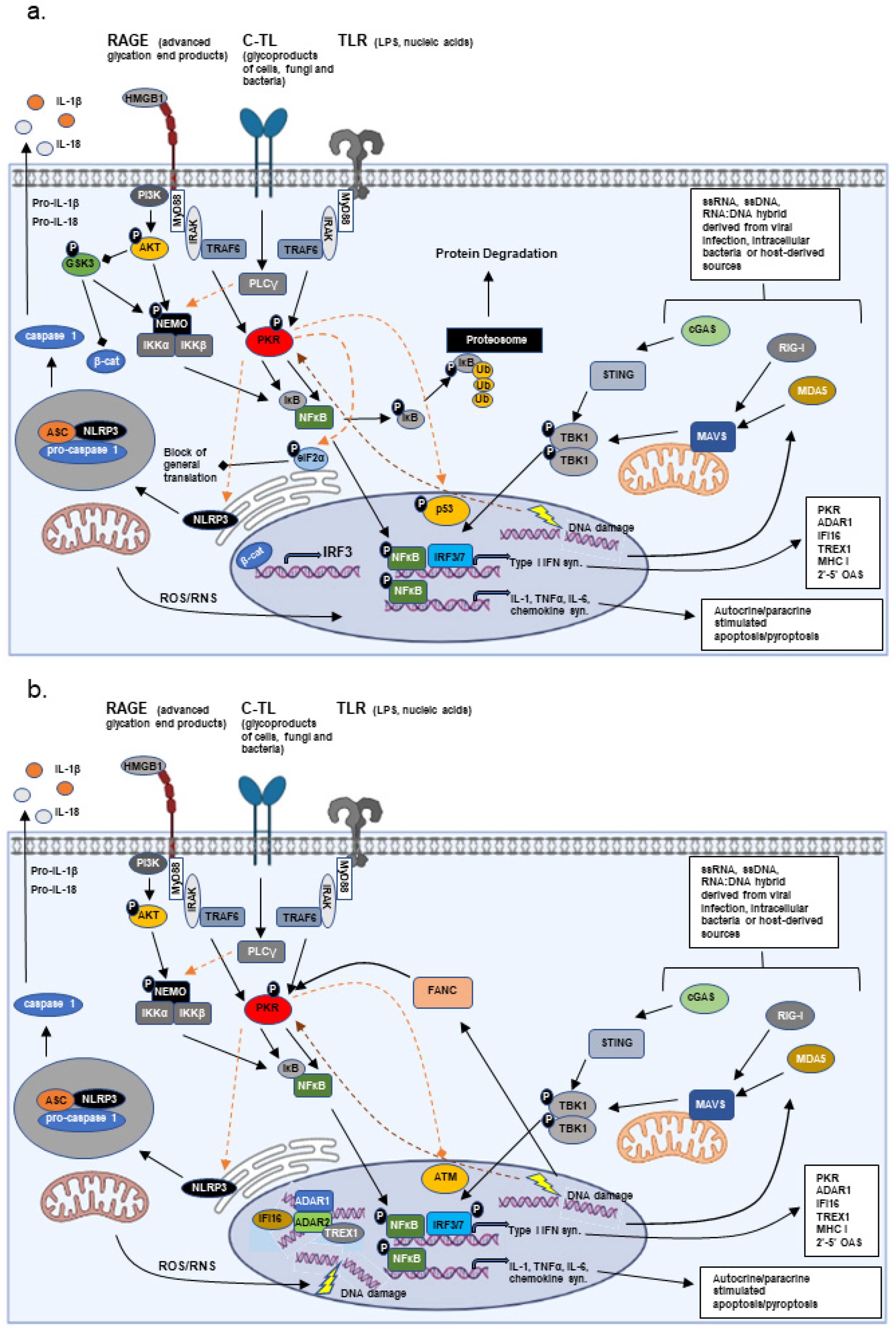
3. Adaption vs. Disease in Chronic Inflammation
2.1. Bone Marrow Failure Disorders and Acute Myelogenous Leukemia
2.2. Hemoglobin Beta
2.3. Gastro-Intestinal Microbiome
2.4. Innate Immune Control of DNA Damage Repair and Epigentic Modifications
References
- Granger, D.N.; Senchenkova, E. Inflammation and the Microcirculation; Morgan & Claypool Life Sciences Publisher: San Rafael, CA, USA, 2010.
- Gasteiger, G.; D’Osualdo, A.; Schubert, D.A.; Weber, A.; Bruscia, E.M.; Hartl, D. Cellular Innate Immunity: An Old Game with New Players. J. Innate Immun. 2017, 9, 111–125.
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820.
- Prantner, D.; Nallar, S.; Vogel, S.N. The role of RAGE in host pathology and crosstalk between RAGE and TLR4 in innate immune signal transduction pathways. FASEB J. 2020, 34, 15659–15674.
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551.
- Gan, Y.; Li, X.; Han, S.; Liang, Q.; Ma, X.; Rong, P.; Wang, W.; Li, W. The cGAS/STING Pathway: A Novel Target for Cancer Therapy. Front. Immunol. 2021, 12, 795401.
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832.
- Gal-Ben-Ari, S.; Barrera, I.; Ehrlich, M.; Rosenblum, K. PKR: A Kinase to Remember. Front. Mol. Neurosci. 2018, 11, 480.
- Ito, T.; Yang, M.; May, W.S. RAX, a cellular activator for double-stranded RNA-dependent protein kinase during stress signaling. J. Biol. Chem. 1999, 274, 15427–15432.
- Patel, R.C.; Sen, G.C. PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO J. 1998, 17, 4379–4390.
- Blalock, W.L. Opposing forces fight over the same ground to regulate interferon signaling. Biochem. J. 2021, 478, 1853–1859.
- Piazzi, M.; Bavelloni, A.; Faenza, I.; Blalock, W. Glycogen synthase kinase (GSK)-3 and the double-strand RNA-dependent kinase, PKR: When two kinases for the common good turn bad. Biochim. Et Biophys. Acta. Mol. Cell Res. 2020, 1867, 118769.
- Lu, B.; Nakamura, T.; Inouye, K.; Li, J.; Tang, Y.; Lundback, P.; Valdes-Ferrer, S.I.; Olofsson, P.S.; Kalb, T.; Roth, J.; et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature 2012, 488, 670–674.
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal Transduct. Target. Ther. 2021, 6, 291.
- Kok, K.H.; Lui, P.Y.; Ng, M.H.; Siu, K.L.; Au, S.W.; Jin, D.Y. The double-stranded RNA-binding protein PACT functions as a cellular activator of RIG-I to facilitate innate antiviral response. Cell Host Microbe 2011, 9, 299–309.
- Bennett, R.L.; Blalock, W.L.; Abtahi, D.M.; Pan, Y.; Moyer, S.A.; May, W.S. RAX, the PKR activator, sensitizes cells to inflammatory cytokines, serum withdrawal, chemotherapy, and viral infection. Blood 2006, 108, 821–829.
- Sokol, C.L.; Luster, A.D. The chemokine system in innate immunity. Cold Spring Harb. Perspect. Biol. 2015, 7, a016303.
- Eiz-Vesper, B.; Schmetzer, H.M. Antigen-Presenting Cells: Potential of Proven und New Players in Immune Therapies. Transfus. Med. Hemother. 2020, 47, 429–431.
- Kambayashi, T.; Laufer, T.M. Atypical MHC class II-expressing antigen-presenting cells: Can anything replace a dendritic cell? Nat. Rev. Immunol. 2014, 14, 719–730.
- Dong, C.; Flavell, R.A. Cell fate decision: T-helper 1 and 2 subsets in immune responses. Arthritis Res. 2000, 2, 179–188.
- Uribe-Querol, E.; Rosales, C. Phagocytosis: Our Current Understanding of a Universal Biological Process. Front. Immunol. 2020, 11, 1066.
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49.
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902.
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, e17023.
- Liu, T.; Zhou, Y.; Ko, K.S.; Yang, H. Interactions between Myc and Mediators of Inflammation in Chronic Liver Diseases. Mediat. Inflamm. 2015, 2015, 276850.
- Bae, S.; Park, P.S.U.; Lee, Y.; Mun, S.H.; Giannopoulou, E.; Fujii, T.; Lee, K.P.; Violante, S.N.; Cross, J.R.; Park-Min, K.H. MYC-mediated early glycolysis negatively regulates proinflammatory responses by controlling IRF4 in inflammatory macrophages. Cell Rep. 2021, 35, 109264.
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Et Biophys. Acta 2014, 1843, 2563–2582.
- Cheroni, C.; Manganaro, L.; Donnici, L.; Bevilacqua, V.; Bonnal, R.J.P.; Rossi, R.L.; De Francesco, R. Novel interferon-sensitive genes unveiled by correlation-driven gene selection and systems biology. Sci. Rep. 2021, 11, 18043.
- Uzhachenko, R.V.; Shanker, A. CD8(+) T Lymphocyte and NK Cell Network: Circuitry in the Cytotoxic Domain of Immunity. Front. Immunol. 2019, 10, 1906.
- Muntjewerff, E.M.; Meesters, L.D.; van den Bogaart, G. Antigen Cross-Presentation by Macrophages. Front. Immunol. 2020, 11, 1276.
- Belizario, J.E.; Brandao, W.; Rossato, C.; Peron, J.P. Thymic and Postthymic Regulation of Naive CD4(+) T-Cell Lineage Fates in Humans and Mice Models. Mediat. Inflamm. 2016, 2016, 9523628.
- Van Niekerk, G.; Davids, L.M.; Hattingh, S.M.; Engelbrecht, A.M. Cancer stem cells: A product of clonal evolution? Int. J. Cancer 2017, 140, 993–999.
- Savage, S.A.; Dufour, C. Classical inherited bone marrow failure syndromes with high risk for myelodysplastic syndrome and acute myelogenous leukemia. Semin. Hematol. 2017, 54, 105–114.
- Blalock, W.L.; Bavelloni, A.; Piazzi, M.; Faenza, I.; Cocco, L. A role for PKR in hematologic malignancies. J. Cell. Physiol. 2010, 223, 572–591.
- Piazzi, M.; Bavelloni, A.; Gallo, A.; Faenza, I.; Blalock, W.L. Signal Transduction in Ribosome Biogenesis: A Recipe to Avoid Disaster. Int. J. Mol. Sci. 2019, 20, 2718.
- Fisher, D.A.C.; Fowles, J.S.; Zhou, A.; Oh, S.T. Inflammatory Pathophysiology as a Contributor to Myeloproliferative Neoplasms. Front. Immunol. 2021, 12, 683401.
- Zhang, X.; Li, J.; Sejas, D.P.; Rathbun, K.R.; Bagby, G.C.; Pang, Q. The Fanconi anemia proteins functionally interact with the protein kinase regulated by RNA (PKR). J. Biol. Chem. 2004, 279, 43910–43919.
- Follo, M.Y.; Finelli, C.; Mongiorgi, S.; Clissa, C.; Bosi, C.; Martinelli, G.; Blalock, W.L.; Cocco, L.; Martelli, A.M. PKR is activated in MDS patients and its subcellular localization depends on disease severity. Leukemia 2008, 22, 2267–2269.
- Blalock, W.L.; Grimaldi, C.; Fala, F.; Follo, M.; Horn, S.; Basecke, J.; Martinelli, G.; Cocco, L.; Martelli, A.M. PKR activity is required for acute leukemic cell maintenance and growth: A role for PKR-mediated phosphatase activity to regulate GSK-3 phosphorylation. J. Cell. Physiol. 2009, 221, 232–241.
- Blalock, W.L.; Bavelloni, A.; Piazzi, M.; Tagliavini, F.; Faenza, I.; Martelli, A.M.; Follo, M.Y.; Cocco, L. Multiple forms of PKR present in the nuclei of acute leukemia cells represent an active kinase that is responsive to stress. Leukemia 2011, 25, 236–245.
- Cheng, X.; Byrne, M.; Brown, K.D.; Konopleva, M.Y.; Kornblau, S.M.; Bennett, R.L.; May, W.S. PKR inhibits the DNA damage response, and is associated with poor survival in AML and accelerated leukemia in NHD13 mice. Blood 2015, 126, 1585–1594.
- Melamed, D.; Nov, Y.; Malik, A.; Yakass, M.B.; Bolotin, E.; Shemer, R.; Hiadzi, E.K.; Skorecki, K.L.; Livnat, A. De novo mutation rates at the single-mutation resolution in a human HBB gene region associated with adaptation and genetic disease. Genome Res. 2022, 32, 488–498.
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506.
- Suppli, M.P.; Bagger, J.I.; Lelouvier, B.; Broha, A.; Demant, M.; Konig, M.J.; Strandberg, C.; Lund, A.; Vilsboll, T.; Knop, F.K. Hepatic microbiome in healthy lean and obese humans. JHEP Rep. 2021, 3, 100299.
- Molinero, N.; Ruiz, L.; Milani, C.; Gutierrez-Diaz, I.; Sanchez, B.; Mangifesta, M.; Segura, J.; Cambero, I.; Campelo, A.B.; Garcia-Bernardo, C.M.; et al. The human gallbladder microbiome is related to the physiological state and the biliary metabolic profile. Microbiome 2019, 7, 100.
- Monroe, J.G.; Srikant, T.; Carbonell-Bejerano, P.; Becker, C.; Lensink, M.; Exposito-Alonso, M.; Klein, M.; Hildebrandt, J.; Neumann, M.; Kliebenstein, D.; et al. Mutation bias reflects natural selection in Arabidopsis thaliana. Nature 2022, 602, 101–105.
- Shiromoto, Y.; Sakurai, M.; Minakuchi, M.; Ariyoshi, K.; Nishikura, K. ADAR1 RNA editing enzyme regulates R-loop formation and genome stability at telomeres in cancer cells. Nat. Commun. 2021, 12, 1654.
- Ka, N.L.; Lim, G.Y.; Hwang, S.; Kim, S.S.; Lee, M.O. IFI16 inhibits DNA repair that potentiates type-I interferon-induced antitumor effects in triple negative breast cancer. Cell Rep. 2021, 37, 110138.
- Schoggins, J.W.; Rice, C.M. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 2011, 1, 519–525.
- Jimeno, S.; Balestra, F.R.; Huertas, P. The Emerging Role of RNA Modifications in DNA Double-Strand Break Repair. Front. Mol. Biosci. 2021, 8, 664872.
- Tao, S.S.; Wu, G.C.; Zhang, Q.; Zhang, T.P.; Leng, R.X.; Pan, H.F.; Ye, D.Q. TREX1 As a Potential Therapeutic Target for Autoimmune and Inflammatory Diseases. Curr. Pharm. Des. 2019, 25, 3239–3247.
- Tassinari, V.; Cesarini, V.; Tomaselli, S.; Ianniello, Z.; Silvestris, D.A.; Ginistrelli, L.C.; Martini, M.; De Angelis, B.; De Luca, G.; Vitiani, L.R.; et al. ADAR1 is a new target of METTL3 and plays a pro-oncogenic role in glioblastoma by an editing-independent mechanism. Genome Biol. 2021, 22, 51.
- Lin, Z.; Hsu, P.J.; Xing, X.; Fang, J.; Lu, Z.; Zou, Q.; Zhang, K.J.; Zhang, X.; Zhou, Y.; Zhang, T.; et al. Mettl3-/Mettl14-mediated mRNA N(6)-methyladenosine modulates murine spermatogenesis. Cell Res. 2017, 27, 1216–1230.
- Zhang, C.; Chen, L.; Peng, D.; Jiang, A.; He, Y.; Zeng, Y.; Xie, C.; Zhou, H.; Luo, X.; Liu, H.; et al. METTL3 and N6-Methyladenosine Promote Homologous Recombination-Mediated Repair of DSBs by Modulating DNA-RNA Hybrid Accumulation. Mol. Cell 2020, 79, 425–442.e427.
- Jimeno, S.; Prados-Carvajal, R.; Fernandez-Avila, M.J.; Silva, S.; Silvestris, D.A.; Endara-Coll, M.; Rodriguez-Real, G.; Domingo-Prim, J.; Mejias-Navarro, F.; Romero-Franco, A.; et al. ADAR-mediated RNA editing of DNA:RNA hybrids is required for DNA double strand break repair. Nat. Commun. 2021, 12, 5512.
- Geffroy, B.; Douhard, M. The Adaptive Sex in Stressful Environments. Trends Ecol. Evol. 2019, 34, 628–640.
- Kishimoto, S.; Uno, M.; Okabe, E.; Nono, M.; Nishida, E. Environmental stresses induce transgenerationally inheritable survival advantages via germline-to-soma communication in Caenorhabditis elegans. Nat. Commun. 2017, 8, 14031.
- Cheng, C.; Kirkpatrick, M. Environmental Plasticity in the Intersexual Correlation and Sex Bias of Gene Expression. J. Hered. 2017, 108, 754–758.
- Sharma, U. Paternal Contributions to Offspring Health: Role of Sperm Small RNAs in Intergenerational Transmission of Epigenetic Information. Front. Cell Dev. Biol. 2019, 7, 215.
- Donkin, I.; Barres, R. Sperm epigenetics and influence of environmental factors. Mol. Metab. 2018, 14, 1–11.
- Marcho, C.; Oluwayiose, O.A.; Pilsner, J.R. The preconception environment and sperm epigenetics. Andrology 2020, 8, 924–942.
- Yang, Q.; Wang, G.; Zhang, F. Role of Peripheral Immune Cells-Mediated Inflammation on the Process of Neurodegenerative Diseases. Front. Immunol. 2020, 11, 582825.
- Sim, K.Y.; Im, K.C.; Park, S.G. The Functional Roles and Applications of Immunoglobulins in Neurodegenerative Disease. Int. J. Mol. Sci. 2020, 21, 5295.
- Piazzi, M.; Bavelloni, A.; Cenni, V.; Faenza, I.; Blalock, W.L. Revisiting the Role of GSK3, A Modulator of Innate Immunity, in Idiopathic Inclusion Body Myositis. Cells 2021, 10, 3255.
- Lin, B.; Goldbach-Mansky, R. Pathogenic insights from genetic causes of autoinflammatory inflammasomopathies and interferonopathies. J. Allergy Clin. Immunol. 2022, 149, 819–832.
- Nazmi, A.; Field, R.H.; Griffin, E.W.; Haugh, O.; Hennessy, E.; Cox, D.; Reis, R.; Tortorelli, L.; Murray, C.L.; Lopez-Rodriguez, A.B.; et al. Chronic neurodegeneration induces type I interferon synthesis via STING, shaping microglial phenotype and accelerating disease progression. Glia 2019, 67, 1254–1276.
- Long, H.Z.; Cheng, Y.; Zhou, Z.W.; Luo, H.Y.; Wen, D.D.; Gao, L.C. PI3K/AKT Signal Pathway: A Target of Natural Products in the Prevention and Treatment of Alzheimer’s Disease and Parkinson’s Disease. Front. Pharmacol. 2021, 12, 648636.


