Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Johanna Arroyave-Ospina | -- | 4230 | 2022-06-02 10:46:48 | | | |
| 2 | Vivi Li | -1 word(s) | 4229 | 2022-06-06 03:35:00 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Arroyave-Ospina, J.; Pedroza-Diaz, J.; , . Oxidative Stress-Induced Senescence during Non-Alcoholic Fatty Liver Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/23693 (accessed on 23 July 2026).
Arroyave-Ospina J, Pedroza-Diaz J, . Oxidative Stress-Induced Senescence during Non-Alcoholic Fatty Liver Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/23693. Accessed July 23, 2026.
Arroyave-Ospina, Johanna, Johanna Pedroza-Diaz, . "Oxidative Stress-Induced Senescence during Non-Alcoholic Fatty Liver Disease" Encyclopedia, https://encyclopedia.pub/entry/23693 (accessed July 23, 2026).
Arroyave-Ospina, J., Pedroza-Diaz, J., & , . (2022, June 02). Oxidative Stress-Induced Senescence during Non-Alcoholic Fatty Liver Disease. In Encyclopedia. https://encyclopedia.pub/entry/23693
Arroyave-Ospina, Johanna, et al. "Oxidative Stress-Induced Senescence during Non-Alcoholic Fatty Liver Disease." Encyclopedia. Web. 02 June, 2022.
Copy Citation
Non-alcoholic fatty liver disease is characterized by disturbed lipid metabolism and increased oxidative stress. These conditions lead to the activation of different cellular response mechanisms, including senescence. Cellular senescence constitutes an important response to injury in the liver. Researches show that chronic oxidative stress can induce senescence, and this might be a driving mechanism for NAFLD progression, aggravating the disturbance of lipid metabolism, organelle dysfunction, pro-inflammatory response and hepatocellular damage. In this context, the modulation of cellular senescence can be beneficial to ameliorate oxidative stress-related damage during NAFLD progression.
oxidative stress
ROS
non-alcoholic liver disease
stress-induced senescence
lipid metabolism
ER stress
mitochondrial dysfunction
1. Introduction
Non-alcoholic fatty liver disease (NAFLD) is a condition in which excessive amounts of lipids accumulate in the liver of individuals with no history of alcohol consumption, viral infection or (chronic) drug intoxication. NAFLD can progress to non-alcoholic steatohepatitis (NASH), a condition characterized by steatosis, inflammation, necrosis and fibrosis. The fibrotic stage can progress to cirrhosis and even hepatocellular carcinoma [1][2]. Among chronic liver diseases, NAFLD is considered the most prevalent worldwide, with its incidence and prevalence rapidly rising. The global prevalence of NAFLD is approximately 25%, with the highest prevalence reported in the Middle East (32%) and South America (31%), followed by Asia (27%), the USA (24%) and Europe (23%), whereas NAFLD is less common in Africa (14%) [3][4].
Oxidative stress (OxS) is an important factor in the pathogenesis of NAFLD [5]. OxS causes lipid and DNA damage, leading to organelle dysfunction, involving the impairment of mitochondrial and ER homeostasis, which may lead to senescence [6]. Senescence is a cellular program that induces prolonged or irreversible cell cycle arrest accompanied by distinct phenotypic alterations, including metabolic reprograming and chromatin remodeling. Senescent cells are characterized by at least two features: (1) resistance to apoptosis and (2) the acquisition of a complex pro-inflammatory secretome: the senescence-associated secretory phenotype or SASP [7]. Cellular senescence is triggered by different mechanisms and both endogenous and exogenous factors can induce premature senescence, e.g., lack of nutrients and/or growth factors, irreversible DNA damage, oncogene overexpression, mitochondrial dysfunction, oxidative stress and ER stress [8].
Several studies demonstrate that OxS is an important mediator of the initiation and progression of cellular senescence in the pathogenesis of chronic liver diseases, including NAFLD [9], and cellular senescence has been proposed to be a driving force of hepatic steatosis and an important contributing factor to NAFLD [10]. In this context, cellular senescence is activated as a cellular stress response to control liver damage and inflammation [11] and it has been demonstrated in both parenchymal as well as non-parenchymal liver cells [12].
2. Role of Senescence in the Development of NAFLD
2.1. Molecular Mechanisms of Cellular Senescence
Cellular senescence can be triggered by DNA damage caused by internal or external stimuli in chromosomes and telomeres. Two important mechanisms of cellular senescence are replicative senescence, mainly linked to telomere shortening during aging, and stress-induced senescence triggered by cellular stress, which is usually associated with DNA damage [13], also called premature senescence. Different stressors induce senescence, and they are often associated with p53 regulation. Some specific factors that determine stress-induced senescence are OxS, mitochondrial dysfunction and ER stress, which can induce irreversible DNA damage and activate the molecular mechanisms of senescence [14]. Stress factors induce DNA damage, in particular DNA double-strand breaks (DSBs), triggering the DNA damage response (DDR). The activation of this pathway leads to the activation of ATM (ataxia telangiectasia mutated) and Rad-3-related protein kinases, and subsequently to the phosphorylation of p53 and the activation of p21, which eventually results in cell cycle arrest. In addition, p21 and p16 inhibit the phosphorylation of the Rb protein (retinoblastoma factor), allowing it to bind to the E2F transcription factor and thus contributing to cell cycle arrest [14]. In summary, different cell signaling pathways related to cellular senescence converge in the activation of p53 and subsequently in the activation of the cyclin-dependent kinase (CDK) inhibitors p16 (INK4A), p15 (INK4B), p21 (WAF1) and p27 (CDKN1B) [15]. P53 levels determine cell fate during stress conditions and increased levels are observed in early senescence, but higher levels (at least two-times higher) are usually observed during apoptosis. Likewise, p53 levels can be stabilized and the maintenance of the senescence phenotype is often associated with p16 activation, when the senescent state is already stablished and it cannot be reversed by inhibiting p53 [16].
The DNA damage response is a well-known activator of SASP. The SASP includes an array of pro-inflammatory factors, mostly NF-kB dependent factors such as IL-6, IL-8, IL-1β, MCP-1 (monocyte chemoattractant protein-1), MCP-2 and MCP-4, growth factors such as HGF (human growth factor) and FGF (fibroblast growth factor), proteases such as MMPs (matrix metalloproteinases) and secreted insoluble proteins/extracellular matrix proteins (ECM). SASP can reinforce senescence-induced cell cycle arrest by autocrine or paracrine mechanisms and can modulate the tissue microenvironment by paracrine pathways [17]. The SASP is induced and regulated by several signaling pathways, leading to the activation of NF-κB and/or CCAAT/enhancer-binding protein-β (C/EBPβ). These pathways include the NOTCH signaling pathway [18], the cGAS–STING pathway [19][20] and the NAD+–NAMPT pathway [21]. Furthermore, the SASP seems to be context dependent and its composition varies depending on the type of stimulus, duration and cell type [14].
2.2. Senescence Features in Liver Disease
Hepatocyte senescence can be induced by various stress conditions, such as metabolic stress, viral infections and oncogenic stimuli and it can also be induced by aging [22]. The senescence of hepatocytes appears to be a hallmark in chronic liver disease independent of the etiology. The normal liver contains a rather constant level of senescent hepatocytes (3–7%), but in chronic liver diseases, this percentage may increase to 50–100% [23].
Chronic liver injury contributes to the continuous generation of senescent cells or “chronic” senescence, aggravating liver dysfunction and tumor progression [9]. Moreover, chronic liver injury induces functional changes in hepatocytes, which, sometimes, can be reversed or progress to cell death, depending on the level of injury [22]. In general, senescent hepatocytes undergo morphological and biochemical changes similar to those already reported as markers of cellular senescence, including a reduced number of mitochondria (decreased biogenesis and/or increased mitophagy) [24], the accumulation of lipofuscin, the increased expression and/or activity of HP1β, p21, p16, p53 and γ-H2AX and elevated SA-β-galactosidase activity [25][26][27]. Hepatocyte senescence also leads to the SASP [28], resulting in the production of pro-inflammatory mediators and alterations in the tissue microenvironment. For instance, TGF-β signaling appears to be involved in the “spreading” of senescence from one cell to another. When TGF-β signaling is inhibited in mice, senescence is impeded, regeneration is accelerated and survival is improved [29]. It has also been shown that IL-1 and IL-6 are capable of inducing senescence in adjacent cells, which is antagonized by the inhibition of their receptors [23].
During liver injury of various etiology, including NAFLD, damage-associated molecular patterns (DAMPs) link inflammation to cell death mechanisms [26]. Likewise, cellular senescence is activated as an endogenous response mechanism in cellular stress conditions. Senescence of various parenchymal and non-parenchymal resident liver cells has been demonstrated in chronic liver diseases [9]. Inflammation plays a crucial role in the development of liver injury due the involvement of circulating and resident immune cells e.g., neutrophils, monocytes and macrophages, together with liver non-parenchymal cells such as Kupffer cells (KCs), liver sinusoidal endothelial cells (LSECs) and hepatic stellate cells (HSCs).
Progression of NAFLD is often accompanied by inflammatory events and clinical studies have shown that progression of the disease is often associated with excessive activation of the innate and the adaptive immune response [30]. Lipotoxicity and organelle dysfunction, mediated by OxS, induce hepatocyte cell death, which is a key trigger of liver inflammation in NAFLD. In addition, KCs are activated by phagocytosis of apoptotic bodies leading to the production of TNF, TRAIL and FAS ligand further promoting hepatocyte apoptosis, liver inflammation and fibrosis [31]. Characterization of antigenic stimuli triggering the adaptive immune response in the liver will also be important to elucidate the role of the adaptive immune system in liver inflammation and damage. As already mentioned, OxS and lipid peroxidation are common features of NAFLD. Oxidized phospholipids and reactive aldehydes produced during lipid peroxidation, such as malondialdehyde, induce hepatic inflammation, but also form antigenic adducts with cellular macromolecules known as OxS-derived epitopes (OSEs) [32]. The involvement of OxS in driving NAFLD-associated immune responses was inferred from observations that elevated titers of anti-OSE IgG were detected in approximately 40% of patients with NAFLD or NASH in two unrelated cohorts [33]. These studies demonstrate that OxS is an important contributing factor in the immune response during progression of NAFLD. Likewise, high titers of anti-OSE IgGs are associated with the severity of lobular inflammation, the prevalence of intrahepatic B cell and/or T cell aggregates and are an independent predictor of fibrosis [32][34].
Moreover, in liver injury conditions neutrophils promote ROS production via enzymatic activation of NADPH oxidase, activation of proteases and synthesis of inflammatory mediators that serve as chemo-attractants of other immune cells such as macrophages [35][36]. KCs are directly involved in the progression of NAFLD, since they contribute importantly to hepatic ROS production and release of fibrogenic cytokines such as TGF-β [37], promoting the activation of hepatic stellate cells via SMAD signaling [38]. Considering the participation of immune cells in NAFLD development, it is relevant to mention that neutrophils can trigger telomere dysfunction via ROS production in an in vivo model of acute liver injury using carbon tetrachloride (CCl4). Besides, it has been found that neutrophils can reduce in vitro cell growth of co-cultured fibroblasts, with elevation of senescence markers and high ROS production [39], suggesting that senescence mediated by inflammatory cells is (partly) ROS dependent. In addition, senescent cells mediate the recruitment of neutrophils, thereby aggravating ROS production and inflammation [39]. Interestingly, inflammatory cells are a hallmark of steatohepatitis in NAFLD and increased ROS production can influence directly and indirectly the activation of liver inflammatory cells, a process that might be mediated by the SASP enhancing liver damage and promoting fibrogenesis during NAFLD.
3. Molecular Mechanisms of Oxidative Stress Contributing to Senescence in NAFLD
3.1. Senescence and Oxidative Stress
The role of OxS in the modulation of senescence and excessive ROS production has been identified as a driving factor of senescence [6]. It has been observed that the induction of ROS production in vitro recapitulated senescence-induced changes in healthy at term placental explants. This was demonstrated to be tightly dependent on p21 expression, which might be regulated by OxS [40][41][42]. Cellular senescence also correlates with OxS-induced damage in various tissues and/or cell populations. For instance, increased levels of 8-hydroxy-2′-deoxyguanosine (8-OHdG), a marker of DNA oxidative damage and ROS production have been found to be associated with senescence in different cell types [43][44][45]. Moreover, the induction of OxS is enough to induce cell cycle arrest, as also demonstrated after exposure of IMR-90 cells to hydrogen peroxide (H2O2), in which long-term growth arrest, morphological changes and the activation of SA-β-galactosidase with increased p21 expression were observed [42]. Experiments with endothelial cells have also demonstrated that ROS production induced by hydrogen peroxide treatment correlates with the induction of senescence [43].
NAFLD development and progression are closely related to OxS and increasing experimental evidence suggests that OxS is a critical factor in the regulation of cellular senescence in NAFLD [44]. In the next section, researchers discuss the mechanisms of OxS-induced senescence in hepatocytes and its role in the progression of NAFLD. The main mechanisms and features of OxS-induced senescence in NAFLD are summarized in Figure 1.
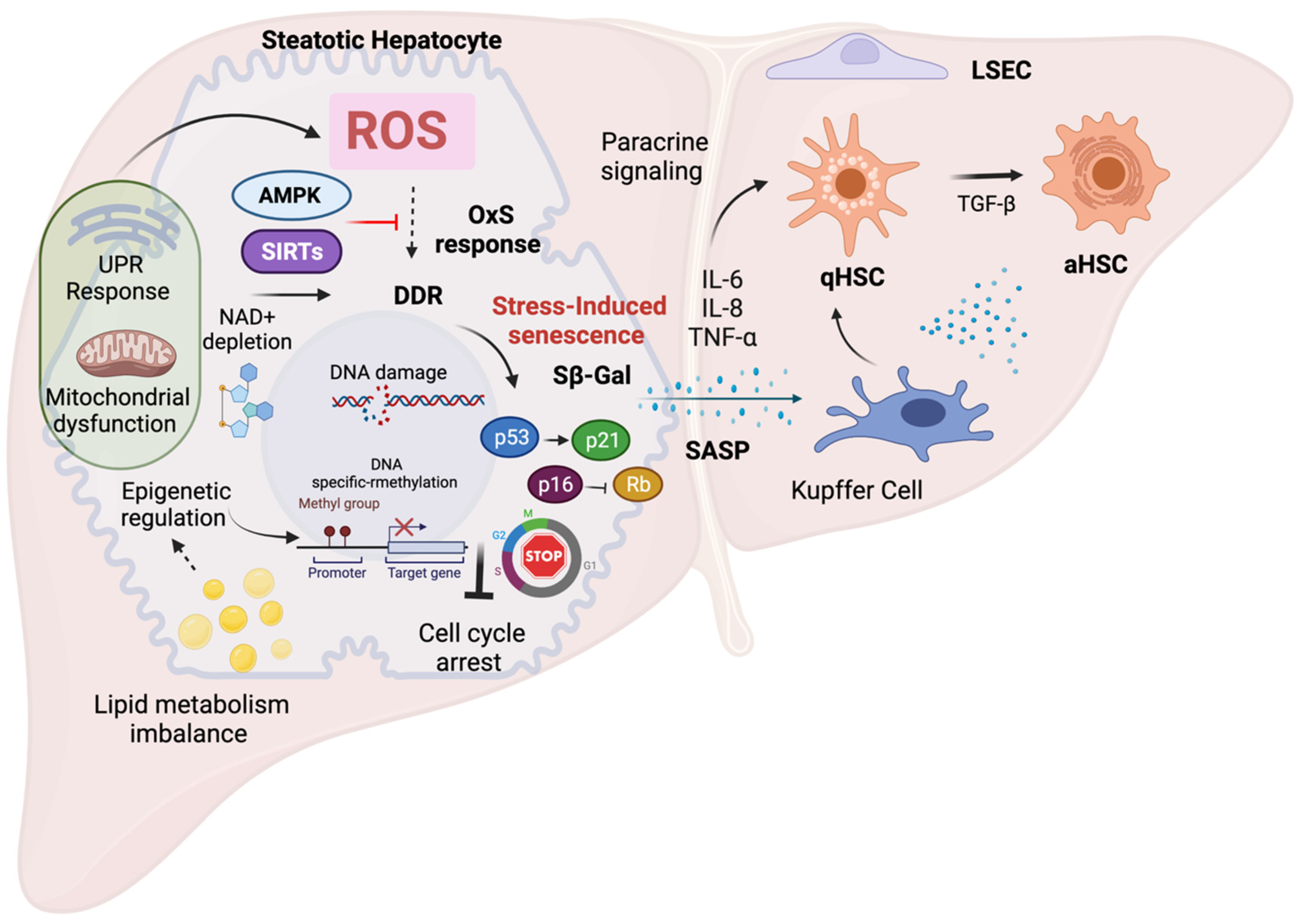
Figure 1. Mechanisms and features of oxidative stress-induced senescenceExcess ROS triggers premature senescence as part of the cellular stress response via activation of the DNA-damage response with concomitant activation of p53-p21 and p16-Rb pathways. This causes prolonged cell cycle arrest and prevents the activation of the cell death program and limits liver damage. Impaired lipid metabolism during NAFLD induces organelle dysfunction, contributing to OxS and senescence. Chronic oxidative stress during NAFLD leads to dysregulation of several factors such as depletion of NAD+ levels with diminished SIRT expression and downregulation of AMPK signaling, leading to deleterious cellular senescence. These pathways can be potential therapeutic targets to control cellular senescence via modulation of OxS. Other cellular response mechanisms such as epigenetic regulation, can directly influence cellular senescence, e.g., via p16 activation. Epigenetic changes have been demonstrated to be induced by lipid accumulation and OxS during NAFLD. Finally, the SASP along with the production of pro-inflammatory factors can influence neighboring cells by paracrine signaling (bystander effect), and allows activation of non-parenchymal cells contributing to NAFLD progression.
In NAFLD, increased ROS production has been linked to the induction of cellular senescence and patterns of gene expression related to senescence. Genome-wide association studies (GWAS) have shown that the p53 (TP53) and p21 genes (Cdkn1a display single-nucleotide polymorphisms (SNPs) in NAFLD patients, resulting in the increased expression of these genes and the inhibition of cell cycle progression as observed in senescent cells [45][46]. Increased p53 expression stimulates p21 expression, which may subsequently increase RB protein phosphorylation and induce continuous activation of the transcription factor E2F1 and increased lipogenesis. When p21 is overexpressed, it inhibits CDK1 function, which may lead to increased OxS, thereby promoting NAFLD. [47]. Therefore, it is plausible to assume that the link between OxS and cellular senescence in NAFLD is bidirectional.
3.2. Senescence and Hepatic Lipid Metabolism
A potential link between senescence and hepatic lipid metabolism has been hypothesized since impaired lipid metabolism is a driving force in NAFLD development and progression and since impaired lipid metabolism is also linked to OxS. Furthermore, steatosis may contribute to DNA damage via OxS and the activation of signaling pathways that lead to cellular senescence [48]. Evidence from in vitro and in vivo findings has shown that the senescent phenotype in hepatocytes correlates with increased fat accumulation [49][50][51]. Likewise, experimental evidence suggests that impaired lipid metabolism can drive cellular senescence, most likely via OxS-dependent mechanisms [52]. Senescent cells also have an altered metabolism during NAFLD development, and because of mitochondrial dysfunction they are less able to metabolize fatty acids, which may contribute to fat accumulation and OxS in the liver [51][53]. Senescent cells show changes in lipid metabolism, but the importance of these changes to cellular senescence is still unclear. Therefore, bidirectional interaction between cellular senescence and lipid metabolism is plausible; however, its causality needs to be further explored.
Transcriptomic and lipidomic analysis has revealed differential expression patterns of lipid-related genes and different lipid compositions in senescent cells, suggesting an important role of lipid metabolism in senescence. Specific polyunsaturated triacylglycerols species accumulate during the development of cellular senescence, probably via increased CD36-mediated fatty acid uptake [54]. Interestingly, a significant increase in CD36 expression was able to induce a senescent-like phenotype, with a concomitant accumulation of phosphatidylcholine. This could be linked to membrane remodeling during the induction of senescence [55]. These findings correlate with the observation that senescent cells accumulate lipid droplets and show increased expression of several lipid regulatory proteins. In addition, in vitro treatment of proliferating cells with specific lipids, such as cholesterol, ceramides or triglycerides, significantly increased cellular senescence [56]. This evidence suggests that the dysregulation of lipid metabolism can induce cellular senescence. Moreover, increased accumulation of lipid droplets in senescent cells during OxS might be a cellular mechanism of protection against lipotoxicity, but this point remains still unclear.
Cellular senescence has also been linked to fatty acid metabolism. Fatty acid composition appears to change during aging and mitochondrial fatty acid oxidation can be affected during senescence. Downregulation of the transcription factor PPARα causes decreased expression of CPT1C, the transmembrane enzyme that mediates fatty acid transport across the mitochondrial membrane and induces cellular senescence in cancer cells [57]. Additional studies have shown that CPT1A upregulation in senescent cells contributes to increased fatty acid oxidation in mitochondria, leading to ROS overproduction and mitochondrial dysfunction. The inhibition of CPT1A reverses these features in senescent cells [58].
Lipid molecules and their metabolites can modulate different cellular responses. Lipids have several biological functions: structural, as part of cellular membranes, in the storage of fatty acids, as neutral lipids in lipid droplets and some lipids, e.g., diacylglycerol, sphingolipids and ceramides, can act as signaling molecules [59]. Sphingolipids and related metabolites such as ceramides, have been proposed as key factors in NAFLD progression because they strongly influence lipid metabolism and lipotoxicity in the liver [60]. In addition, ceramides have been shown to induce cellular senescence in vitro and it has been proposed that ceramides promote senescence during the development of NAFLD [61][62]. The modulation of signaling pathways related to cellular senescence can influence lipid accumulation [12]. However, this modulation is reciprocal, as lipid species are also modulators of cellular senescence and can either be protective or contribute to the progression of NAFLD via ER stress and mitochondrial dysfunction [63].
In summary, current evidence shows that metabolic dysregulation favors cellular senescence. In particular, lipid metabolism appears to play an important role in cellular senescence. In NAFLD, there is disturbed lipid metabolism, which is closely related to OxS and there is also a strong correlation between OxS, lipid metabolism and cellular senescence [64]. Therefore, targeting cellular senescence can have a beneficial impact on lipid metabolism, and the deleterious effects of OxS induced by aberrant lipid accumulation observed in NAFLD conditions.
4. Modulating OxS-Induced Senescence as a Potential Therapy in NAFLD
4.1. Targeting Oxidative Stress to Modulate Cellular Senescence
OxS might be an interesting therapeutic target to modulate senescence. Results from various experimental models have demonstrated that the inhibition of OxS can effectively inhibit cellular senescence [65][66][67][68]. The same has been shown in the aging liver: a number of in vivo studies have demonstrated the attenuation of hepatic senescence by preventing ROS production or by enhancing the antioxidant response [69][70][71]. Nrf2 activation might also attenuate cellular senescence [72], and Nrf2 activators are able to antagonize cellular senescence in human mesenchymal cells [73][74]. In fact, it has been found that activation of Nrf2 with specific compounds suppress cellular senescence in mouse livers, with inhibition of the SASP and DDR pathway and with concomitant reduction of ROS production via upregulation of antioxidant enzymes. In the study, the crucial role of ROS in the induction and maintenance of senescence in the liver as well as the potential as a therapeutic target were convincingly demonstrated [75]. Likewise, the induction of Nrf2 activation in rodents inhibits senescence and the SASP and protects against oxidative damage in different tissues. Interestingly, in the study Nrf2 activation was found to be AMPK-dependent [68].
Findings in a hamster model of vitamin D deficiency demonstrated that vitamin D has anti-senescence effects, which are mediated by Nrf2 activation and subsequent reduction of ROS production [66]. Other experimental results from animal models of NAFLD induced by HFD, demonstrated that vitamin D deficiency was associated with increased cellular senescence and vitamin D supplementation normalized biochemical parameters. Vitamin D supplementation correlated with increased level of SMP-30, a senescence biomarker that is usually downregulated in NAFLD [76]. These results imply that the beneficial antioxidant effects of vitamin D are associated with its potential as a senescence modulator and might be useful in the treatment of NAFLD.
NAD+ metabolism, AMPK and SIRT proteins have been identified as potential molecular targets of the beneficial effects of OxS reduction on cellular senescence [77]. Restoring cellular NAD+ levels protects against senescence and eliminating senescent cells or antagonizing the SASP improves NAD+ homeostasis [78]. Similar findings have been demonstrated with paricalcitol, an agonist of vitamin D, which is able to reduce OxS-induced senescence in bile duct ligated mice. A decreased number of SA-β-gal-positive cells and reduced expression of senescence markers such as p53, p21 and p16 were also observed. The effects of paricalcitol on cellular senescence were due to the inhibition of OxS. Paricalcitol effectively prevented downregulation of SIRT1 expression in bile duct ligated mice and in biliary epithelial cells treated with t-BHP (tert-butyl hydroperoxide), confirming the protective role of the SIRT1 pathway against ROS-induced damage [79]. The protective role of SIRT1 against OxS has also been demonstrated in relation to the beneficial effects of resveratrol [80][81], a well-known antioxidant compound. Resveratrol improved mitochondrial function and biogenesis, increased NAD+ levels and induced AMPK activation in a SIRT1-dependent manner, demonstrating that SIRT1 plays an important role in AMPK activation [80]. Several studies reported that resveratrol protects against OxS and prevents hepatic steatosis in experimental models [82][83], and its antioxidant effects are linked to therapeutic targets such as SIRTs, AMPK and Nrf2, which might be also be involved in the regulation of senescence [84][85].
SIRTs have been proposed as a molecular target for the treatment of NAFLD due to their regulatory role in hepatic metabolism, inflammation and cellular senescence [86]. Studies in SIRT1 knockout mice have confirmed that SIRT1 limits hepatic lipid accumulation and hepatic oxidative stress, which especially prevents mitochondrial ROS production. Thus, SIRT1 seems to play a protective role during the progression of NAFLD [87][88]. Naringenin, a citrus flavonoid, has been found to decrease ROS production and improve the liver antioxidant response via the activation of SIRT1, and to attenuate NAFLD progression in a mouse model [89]. Similar findings have been reported with the alkaloid Berberine. This compound was found to reduce lipid accumulation and steatosis via activation of SIRT3 in a dietary mouse model of NAFLD [90]. Various other antioxidants have been reported to reduce OxS via SIRTs in NAFLD models, including resveratrol, dietary polyphenols and melatonin [91][92][93][94][95].
AMPK activation attenuates hepatic OxS and steatosis in NAFLD models [96][97][98]. AMPK activation also modulates cellular senescence [99]. For instance, AMPK activation via dietary modulation or exercise ameliorates NAFLD and decreases hepatic senescence markers in mice models of NAFLD. The beneficial effects of AMPK activation have (partially) been attributed to increased autophagy and lipophagy [100][101]. Recent in vitro and in vivo results have shown that AMPK activators, e.g., licochalcone D, a compound found in a Chinese licorice herb, reduced OxS-induced senescence by triggering AMPK-mediated autophagy [97]. This demonstrates that AMPK is an interesting signaling hub connecting OxS-induced senescence and NAFLD pathophysiology, implying that AMPK activators with demonstrated inhibitory effects on cellular senescence are potential targets to treat NAFLD [102]. In this regard, it is interesting to note that metformin is known as an AMPK activator with hepatoprotective effects. Metformin has been proposed as a useful therapy in NAFLD and has also been reported as a potential senostatic, as discussed below. Previous results from researchers' group have demonstrated that metformin protects against oxidative stress [103], palmitate-induced lipotoxicity [104] and diclofenac-induced liver toxicity in primary rat hepatocytes [105]. Most of these effects are related to the inhibition of mitochondrial ROS production and the preservation of mitochondrial function. Interestingly, metformin has been reported to inhibit cellular senescence [106]. Moreover, results in human adipose stromal cells demonstrated that metformin attenuated oxidative-stress-induced senescence and improved lipid metabolism and adipocyte function. These effects appeared to be mediated by AMPK activation [107]. Further studies should be conducted to confirm whether the observed hepatoprotective effects of metformin are related to cellular senescence.
4.2. SASP Inhibition: Senostatic/Senomorphic Drugs
Senostatics suppress (part of) the senescence phenotype in cells without causing cell death. Ideally, these drugs should suppress the characteristics of senescent cells that contribute to paracrine tissue damage, i.e., the SASP and ROS production. According to their mechanisms of action, senostatics are classified into two groups: generalized senostatics and precision senostatics. The first group modulates the SASP, while the latter inhibits a specific component of the secretome [108]. The limitations related to these drugs are due to the existence of multiple SASP targets, some of which have essential functions besides senescence, which prohibits using them as targets. Moreover, the high variability of the SASP between tissues and during disease development might make it difficult to implement this type of therapy [109][110].
Different senostatic drugs have been proposed for therapeutic application. For instance, rapamycin, a drug targeting mTOR signaling, can reduce the rate of aging. It is known that the mTOR pathway is related to the detrimental effects of aging and its inhibition could improve age-related diseases, lifespan and health span [111]. The anti-aging effect of rapamycin was reported in a study using old mice (aged 25 months) with a 21-month dietary treatment with rapamycin. Transcriptome and pathway analysis showed that the pathway related to mitochondrial function was the most significantly altered one, among a total of 13 significantly altered pathways in the rapamycin-treated group [112]. Likewise, rapamycin has been shown to reduce mitochondrial ROS production [113], and to improve liver function and decrease fibrosis in a rat model of cirrhosis [114]. Similar beneficial findings were reported in mice treated with rapamycin (14 mg/kg diet for 7 weeks) and middle-aged mice: rapamycin attenuated severe age-induced damage to mitochondria, including ROS production, accumulation of mitochondrial DNA fragments, mitochondrial lipoperoxidation and lipofuscin accumulation [115]. In this regard, the modulation of autophagy using a mTOR inhibitor has been proposed as an effective way to modulate cellular senescence [116], and suggests that this is an interesting novel therapeutic target in NAFLD, that might connect OxS and senescence mechanisms.
Other drugs have been shown to have senostatic effects on the liver, most likely via inhibition of the SASP. It has been demonstrated that metformin inhibits the expression of several pro-inflammatory cytokines that are part of the SASP. It also prevents the translocation of NF-κB to the nucleus and inhibits the phosphorylation of IκB and IKKα/β, thus inhibiting the NF-κB pathway [117][118]. In the context of NAFLD, a number of studies have demonstrated that metformin reverses steatosis in murine models with NAFLD [119], NASH [120] and in patients with NAFLD [121]. Glucocorticoids have also been proposed as potential senostatics, due to their inhibitory effects on the SASP, although their effect in chronic liver disease has not been well established [122]. Finally, several other molecules have been proposed as senostatics, such as JAK inhibitors, JNK inhibitors, HDAC inhibitors and small molecule MDM2 antagonists. These molecules modulate (part of) the SASP, enhance mitochondrial activity and reduce cytoplasmic chromatin fragments in senescent cells [123][124][125].
References
- Kleiner, D.E.; Makhlouf, H.R.; Program, D.; Investigation, P.; Branch, R.; Shams, A. Histology of NAFLD and NASH in Adults and Children. Clin. Liver Dis. 2017, 20, 293–312.
- Yasui, K.; Hashimoto, E.; Komorizono, Y.; Koike, K.; Arii, S.; Imai, Y.; Shima, T.; Kanbara, Y.; Saibara, T.; Mori, T.; et al. Characteristics of Patients With Nonalcoholic Steatohepatitis Who Develop Hepatocellular Carcinoma. Clin. Gastroenterol. Hepatol. 2011, 9, 428–433.
- Huang, D.Q.; El-Serag, H.B.; Loomba, R. Global epidemiology of NAFLD-related HCC: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 223–238.
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84.
- Spahis, S.; Delvin, E.; Borys, J.M.; Levy, E. Oxidative Stress as a Critical Factor in Nonalcoholic Fatty Liver Disease Pathogenesis. Antioxid. Redox Signal. 2017, 26, 519–541.
- Pole, A.; Dimri, M.; Dimri, G.P. Oxidative stress, cellular senescence and ageing. AIMS Mol. Sci. 2016, 3, 300–324.
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular senescence and its effector programs. Genes Dev. 2014, 28, 99–114.
- Ben-Porath, I.; Weinberg, R.A. The signals and pathways activating cellular senescence. Int. J. Biochem. Cell Biol. 2005, 37, 961–976.
- Aravinthan, A.D.; Alexander, G.J.M. Senescence in chronic liver disease: Is the future in aging? J. Hepatol. 2016, 65, 825–834.
- Meijnikman, A.S.; Herrema, H.; Scheithauer, T.P.M.; Kroon, J.; Nieuwdorp, M.; Groen, A.K. Evaluating causality of cellular senescence in non-alcoholic fatty liver disease. JHEP Rep. 2021, 3, 100301.
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246.
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453.
- Wood, N.E.; Kositangool, P.; Hariri, H.; Marchand, A.J.; Henne, W.M. Nutrient Signaling, Stress Response, and Inter-organelle Communication Are Non-canonical Determinants of Cell Fate. Cell Rep. 2020, 33, 108446.
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 485.
- Chicas, A.; Wang, X.; Zhang, C.; McCurrach, M.; Zhao, Z.; Mert, O.; Dickins, R.A.; Narita, M.; Zhang, M.; Lowe, S.W. Dissecting the Unique Role of the Retinoblastoma Tumor Suppressor during Cellular Senescence. Cancer Cell 2010, 17, 376–387.
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of p53 in the regulation of cellular senescence. Biomolecules 2020, 10, 420.
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479.
- Hoare, M.; Ito, Y.; Kang, T.W.; Weekes, M.P.; Matheson, N.J.; Patten, D.A.; Shetty, S.; Parry, A.J.; Menon, S.; Salama, R.; et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat. Cell Biol. 2016, 18, 979–992.
- Glück, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 2017, 19, 1061–1070.
- Li, T.; Chen, Z.J. The cGAS-cGAMP-STI NG pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 2018, 215, 1287–1299.
- Nacarelli, T.; Lau, L.; Fukumoto, T.; Zundell, J.; Fatkhutdinov, N.; Wu, S.; Aird, K.M.; Iwasaki, O.; Kossenkov, A.V.; Schultz, D.; et al. NAD + metabolism governs the proinflammatory senescence-associated secretome. Nat. Cell Biol. 2019, 21, 397–407.
- Xiao, M.; Chen, W.; Wang, C.; Wu, Y.; Zhu, S.; Zeng, C.; Cai, Y.; Liu, C.; He, Z. Senescence and cell death in chronic liver injury: Roles and mechanisms underlying hepatocarcinogenesis. Oncotarget 2018, 9, 8772–8784.
- Huda, N.; Liu, G.; Hong, H.; Yan, S.; Khambu, B.; Yin, X.M. Hepatic senescence, the good and the bad. World J. Gastroenterol. 2019, 25, 5069–5081.
- Dabravolski, S.A.; Bezsonov, E.E.; Orekhov, A.N. The role of mitochondria dysfunction and hepatic senescence in NAFLD development and progression. Biomed. Pharmacother. 2021, 142, 112041.
- Irvine, K.M.; Skoien, R.; Bokil, N.J.; Melino, M.; Thomas, G.P.; Loo, D.; Gabrielli, B.; Hill, M.M.; Sweet, M.J.; Clouston, A.D.; et al. Senescent human hepatocytes express a unique secretory phenotype and promote macrophage migration. World J. Gastroenterol. 2014, 20, 17851–17862.
- Hunt, N.J.; Kang, S.W.S.; Lockwood, G.P.; Le Couteur, D.G.; Cogger, V.C. Hallmarks of Aging in the Liver. Comput. Struct. Biotechnol. J. 2019, 17, 1151–1161.
- Wang, M.J.; Chen, F.; Li, J.X.; Liu, C.C.; Zhang, H.B.; Xia, Y.; Yu, B.; You, P.; Xiang, D.; Lu, L.; et al. Reversal of hepatocyte senescence after continuous in vivo cell proliferation. Hepatology 2014, 60, 349–361.
- Campisi, J.; D’Adda Di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740.
- Bird, T.G.; Mller, M.; Boulter, L.; Vincent, D.F.; Ridgway, R.A.; Lopez-Guadamillas, E.; Lu, W.Y.; Jamieson, T.; Govaere, O.; Campbell, A.D.; et al. TGFβ inhibition restores a regenerative response in acute liver injury by suppressing paracrine senescence. Sci. Transl. Med. 2018, 10, eaan1230.
- Wang, H.; Mehal, W.; Nagy, L.E.; Rotman, Y. Immunological mechanisms and therapeutic targets of fatty liver diseases. Cell. Mol. Immunol. 2021, 18, 73–91.
- Canbay, A.; Feldstein, A.E.; Higuchi, H.; Werneburg, N.; Grambihler, A.; Bronk, S.F.; Gores, G.J. Kupffer Cell Engulfment of Apoptotic Bodies Stimulates Death Ligand and Cytokine Expression. Hepatology 2003, 38, 1188–1198.
- Sutti, S.; Albano, E. Adaptive immunity: An emerging player in the progression of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 81–92.
- Busch, C.J.; Hendrikx, T.; Weismann, D.; Jäckel, S.; Sofie, M.; Walenbergh, A.; Rendeiro, A.F.; Weißer, J.; Puhm, F.; Göderle, L.; et al. Malondialdehyde epitopes are sterile mediators of hepatic inflammation in hypercholesterolemic mice. Hepatology 2018, 65, 1181–1195.
- Pitzalis, C.; Jones, G.W.; Bombardieri, M.; Jones, S.A. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat. Rev. Immunol. 2014, 14, 447–462.
- Gao, B.; Tsukamato, H. Inflammation in alcoholic and nonalcoholic fatty liver disease: Friend or foe? Gastroenterology 2016, 150, 1704–1709.
- Smirnov, A.; Daily, K.P.; Criss, A.K. Assembly of NADPH oxidase in human neutrophils is modulated by the opacity-associated protein expression state of Neisseria gonorrhoeae. Infect. Immun. 2014, 82, 1036–1044.
- Meng, F.; Wang, K.; Aoyama, T.; Grivennikov, S.; Paik, Y.; Scholten, D.; Cong, M.; Iwaisako, K.; Liu, X.; Zhang, M.; et al. IL-17 signaling in inflammatory cells, Kupffer cells and Hepatic Stellate cells exacerbates liver fibrosis. Gastroenterology 2012, 143, 765–776.
- Dooley, S.; Ten Dijke, P. TGF-β in progression of liver disease. Cell Tissue Res. 2012, 347, 245–256.
- Lagnado, A.; Leslie, J.; Ruchaud-Sparagano, M.; Victorelli, S.; Hirsova, P.; Ogrodnik, M.; Collins, A.L.; Vizioli, M.G.; Habiballa, L.; Saretzki, G.; et al. Neutrophils induce paracrine telomere dysfunction and senescence in ROS-dependent manner. EMBO J. 2021, 40, e106048.
- Cindrova-Davies, T.; Fogarty, N.M.E.; Jones, C.J.P.; Kingdom, J.; Burton, G.J. Evidence of oxidative stress-induced senescence in mature, post-mature and pathological human placentas. Placenta 2018, 68, 15–22.
- Saroyo, Y.B.; Wibowo, N.; Irwinda, R.; Prijanti, A.R.; Yunihastuti, E.; Bardosono, S.; Krisnadi, S.R.; Permata, P.I.; Wijaya, S.; Santawi, V.P.A. Oxidative Stress Induced Damage and Early Senescence in Preterm Placenta. J. Pregnancy 2021, 2021, 9923761.
- Chen, Q.M.; Prowse, K.R.; Tu, V.C.; Purdom, S.; Linskens, M.H.K. Uncoupling the senescent phenotype from telomere shortening in hydrogen peroxide-treated fibroblasts. Exp. Cell Res. 2001, 265, 294–303.
- Xiao, X.; Xu, M.; Yu, H.; Wang, L.; Li, X.; Rak, J.; Wang, S.; Zhao, R.C. Mesenchymal stem cell-derived small extracellular vesicles mitigate oxidative stress-induced senescence in endothelial cells via regulation of miR-146a/Src. Signal Transduct. Target. Ther. 2021, 6, 354.
- Delli Bovi, A.P.; Marciano, F.; Mandato, C.; Siano, M.A.; Savoia, M.; Vajro, P. Oxidative Stress in Non-alcoholic Fatty Liver Disease. An Updated Mini Review. Front. Med. 2021, 8, 165.
- Kung, C.; Leu, J.I.; Basu, S.; Khaku, S.; Anokye-, F.; Liu, Q.; George, D.L.; Ahima, R.S.; Murphy, M.E. The P72R polymorphism of p53 predisposes to obesity and metabolic dysfunction. Cell Rep. 2016, 14, 2413–2425.
- Aravinthan, A.; Mells, G.; Allison, M.; Leathart, J.; Kotronen, A.; Yki-Jarvinen, H.; Daly, A.K.; Day, C.P.; Anstee, Q.M.; Alexander, G. Gene polymorphisms of cellular senescence marker p21 and disease progression in non-alcohol-related fatty liver disease. Cell Cycle 2014, 13, 1489–1494.
- Caldez, M.J.; Bjorklund, M.; Kaldis, P. Cell cycle regulation in NAFLD: When imbalanced metabolism limits cell division. Hepatol. Int. 2020, 14, 463–474.
- Rudolph, K.L.; Chang, S.; Millard, M.; Schreiber-Agus, N.; DePinho, R.A. Inhibition of experimental liver cirrhosis in mice by telomerase gene delivery. Science 2000, 287, 1253–1258.
- Moustakas, I.I.; Katsarou, A.; Legaki, A.I.; Pyrina, I.; Ntostoglou, K.; Papatheodoridi, A.M.; Gercken, B.; Pateras, I.S.; Gorgoulis, V.G.; Koutsilieris, M.; et al. Hepatic senescence accompanies the development of NAFLD in non-aged mice independently of obesity. Int. J. Mol. Sci. 2021, 22, 3446.
- Zhang, J.; Li, Y.; Wang, B.; Luo, Y.; Shi, J.; Zhao, B. The p66shc-mediated regulation of hepatocyte senescence influences hepatic steatosis in nonalcoholic fatty liver disease. Med. Sci. Monit. 2020, 26, e921887-1.
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 2017, 8, 70–72.
- Chen, Q.; Tang, L.; Xin, G.; Li, S.; Ma, L.; Xu, Y.; Zhuang, M.; Xiong, Q.; Wei, Z.; Xing, Z.; et al. Oxidative stress mediated by lipid metabolism contributes to high glucose-induced senescence in retinal pigment epithelium. Free Radic. Biol. Med. 2019, 130, 48–58.
- Ogrodnik, M.; Zhu, Y.; Langhi, L.G.P.; Tchkonia, T.; Krüger, P.; Fielder, E.; Victorelli, S.; Ruswhandi, R.A.; Giorgadze, N.; Pirtskhalava, T.; et al. Obesity-Induced Cellular Senescence Drives Anxiety and Impairs Neurogenesis. Cell Metab. 2019, 29, 1233.
- Lizardo, D.Y.; Lin, Y.L.; Gokcumen, O.; Atilla-Gokcumen, G.E. Regulation of lipids is central to replicative senescence. Mol. Biosyst. 2017, 13, 498–509.
- Saitou, M.; Lizardo, D.Y.; Taskent, R.O.; Millner, A.; Gokcumen, O.; Atilla-Gokcumen, G.E. An evolutionary transcriptomics approach links CD36 to membrane remodeling in replicative senescence. Mol. Omi. 2018, 14, 237–246.
- Flor, A.C.; Wolfgeher, D.; Wu, D.; Kron, S.J. A signature of enhanced lipid metabolism, lipid peroxidation and aldehyde stress in therapy-induced senescence. Cell Death Discov. 2017, 3, 17075.
- Chen, Y.; Wang, Y.; Huang, Y.; Zeng, H.; Hu, B.; Guan, L.; Zhang, H.; Yu, A.M.; Johnson, C.H.; J.Gonzalez, F.; et al. PPARα regulates tumor cell proliferation and senescence via a novel target gene carnitine palmitoyltransferase 1C. Carcinogenesis 2017, 38, 474–483.
- Seok, J.; Jung, H.S.; Park, S.; Lee, J.O.; Kim, C.J.; Kim, G.J. Alteration of fatty acid oxidation by increased CPT1A on replicative senescence of placenta-derived mesenchymal stem cells. Stem Cell Res. Ther. 2020, 11, 1.
- Millner, A.; Ekin Atilla-Gokcumen, G. Lipid players of cellular senescence. Metabolites 2020, 10, 339.
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191.
- Palmer, A.K.; Tchkonia, T.; LeBrasseur, N.K.; Chini, E.N.; Xu, M.; Kirkland, J.L. Cellular Senescence in Type 2 Diabetes: A Therapeutic Opportunity. Diabetes 2015, 64, 2289–2298.
- Nikolova-Karakashian, M. Sphingolipids at the Crossroads of NAFLD and Senescence. Adv. Cancer Res. 2018, 140, 155–190.
- Lebeaupin, C.; Vallée, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 927–947.
- Papatheodoridi, A.M.; Chrysavgis, L.; Koutsilieris, M.; Chatzigeorgiou, A. The Role of Senescence in the Development of Nonalcoholic Fatty Liver Disease and Progression to Nonalcoholic Steatohepatitis. Hepatology 2020, 71, 363–374.
- Lu, D.; Le, Y.; Ding, J.; Dou, X.; Mao, W.; Zhu, J.; Dou, X.; Mao, W.; Zhu, J.; Dou, X.; et al. CLIC1 Inhibition Protects against Cellular Senescence and Endothelial Dysfunction Via the Nrf2_HO-1 Pathway.pdf. Cell Biochem. Biophys. 2021, 79, 239–252.
- Chen, L.; Yang, R.; Qiao, W.; Zhang, W.; Chen, J.; Mao, L.; Goltzman, D.; Miao, D. 1,25-Dihydroxyvitamin D exerts an antiaging role by activation of Nrf2-antioxidant signaling and inactivation of p16/p53-senescence signaling. Aging Cell 2019, 18, e12951.
- Jie, Z.; Huan, Y.; Mengyun, W.; Yasha, L.; Huafeng, P.; Ke, Y. Nrf2 modulates immunosuppressive ability and cellular senescence of human umbilical cord mesenchymal stem cells. Biochem. Biophys. Res. Commun. 2020, 526, 1021–1027.
- Wang, Z.; Chen, Z.; Jiang, Z.; Luo, P.; Liu, L.; Huang, Y.; Wang, H.; Wang, Y.; Long, L.; Tan, X.; et al. Cordycepin prevents radiation ulcer by inhibiting cell senescence via NRF2 and AMPK in rodents. Nat. Commun. 2019, 10, 2538.
- Zhang, X.; Wu, J.Z.; Lin, Z.X.; Yuan, Q.J.; Li, Y.C.; Liang, J.L.; Zhan, J.Y.X.; Xie, Y.L.; Su, Z.R.; Liu, Y.H. Ameliorative effect of supercritical fluid extract of Chrysanthemum indicum Linnén against D-galactose induced brain and liver injury in senescent mice via suppression of oxidative stress, inflammation and apoptosis. J. Ethnopharmacol. 2019, 234, 44–56.
- Zeng, L.; Lin, L.; Peng, Y.; Yuan, D.; Zhang, S.; Gong, Z.; Xiao, W. L-Theanine attenuates liver aging by inhibiting advanced glycation end products in D-galactose-induced rats and reversing an imbalance of oxidative stress and inflammation. Exp. Gerontol. 2020, 131, 110823.
- Wu, J.L.; Wu, Q.P.; Yang, X.F.; Wei, M.K.; Zhang, J.M.; Huang, Q.; Zhou, X.Y. L-malate reverses oxidative stress and antioxidative defenses in liver and heart of aged rats. Physiol. Res. 2008, 57, 261–268.
- Ungvari, Z.; Tarantini, S.; Nyúl-Tóth, Á.; Kiss, T.; Yabluchanskiy, A.; Csipo, T.; Balasubramanian, P.; Lipecz, A.; Benyo, Z.; Csiszar, A. Nrf2 dysfunction and impaired cellular resilience to oxidative stressors in the aged vasculature: From increased cellular senescence to the pathogenesis of age-related vascular diseases. GeroScience 2019, 41, 727–738.
- Shin, J.H.; Jeon, H.J.; Park, J.; Chang, M.S. Epigallocatechin-3-gallate prevents oxidative stress-induced cellular senescence in human mesenchymal stem cells via Nrf2. Int. J. Mol. Med. 2016, 38, 1075–1082.
- Fang, J.; Yan, Y.; Teng, X.; Wen, X.; Li, N.; Peng, S.; Liu, W.; Donadeu, F.X.; Zhao, S.; Hua, J. Melatonin prevents senescence of canine adipose-derived mesenchymal stem cells through activating NRF2 and inhibiting ER stress. Aging 2018, 10, 2954–2972.
- Gao, L.B.; Wang, Y.H.; Liu, Z.H.; Sun, Y.; Cai, P.; Jing, Q. Identification of a small molecule SR9009 that activates NRF2 to counteract cellular senescence. Aging Cell 2021, 20, e13483.
- Al-ghamdi, H.A.; Al Fayez, F.F.; Bima, A.I.; Khawaji, T.M.; Elsamanoudy, A.Z. Study of Cellular Senescence and Vitamin D Deficiency in Nonalcoholic Fatty Liver Disease and The Potential Protective Effect of Vitamin D Supplementation. J. Clin. Exp. Hepatol. 2021, 11, 219–226.
- Katsyuba, E.; Mottis, A.; Zietak, M.; De Franco, F.; van der Velpen, V.; Gariani, K.; Ryu, D.; Cialabrini, L.; Matilainen, O.; Liscio, P.; et al. De novo NAD+ synthesis enhances mitochondrial function and improves health. Nature 2018, 563, 354–359.
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD+ metabolism: Pathophysiologic mechanisms and therapeutic potential. Signal Transduct. Target. Ther. 2020, 5, 227.
- Jia, R.; Yang, F.; Yan, P.; Ma, L.; Yang, L.; Li, L. Paricalcitol inhibits oxidative stress-induced cell senescence of the bile duct epithelium dependent on modulating Sirt1 pathway in cholestatic mice. Free Radic. Biol. Med. 2021, 169, 158–168.
- Price, N.L.; Gomes, A.P.; Ling, A.J.Y.; Duarte, F.V.; Martin-Montalvo, A.; North, B.J.; Agarwal, B.; Ye, L.; Ramadori, G.; Teodoro, J.S.; et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012, 15, 675–690.
- Ido, Y.; Duranton, A.; Lan, F.; Weikel, K.A.; Breton, L.; Ruderman, N.B. Resveratrol prevents oxidative stress-induced senescence and proliferative dysfunction by activating the AMPK-FOXO3 cascade in cultured primary human keratinocytes. PLoS ONE 2015, 10, e0115341.
- Izdebska, M.; Piątkowska-Chmiel, I.; Korolczuk, A.; Herbet, M.; Gawrońska-Grzywacz, M.; Gieroba, R.; Sysa, M.; Czajkowska-Bania, K.; Cygal, M.; Korga, A.; et al. The beneficial effects of resveratrol on steatosis and mitochondrial oxidative stress in HepG2 cells. Can. J. Physiol. Pharmacol. 2017, 95, 1442–1453.
- Bujanda, L.; Hijona, E.; Larzabal, M.; Beraza, M.; Aldazabal, P.; García-Urkia, N.; Sarasqueta, C.; Cosme, A.; Irastorza, B.; González, A.; et al. Resveratrol inhibits nonalcoholic fatty liver disease in rats. BMC Gastroenterol. 2008, 8, 40.
- Faghihzadeh, F.; Hekmatdoost, A.; Adibi, P. Resveratrol and liver: A systematic review. J. Res. Med. Sci. 2015, 20, 797–810.
- Izzo, C.; Annunziata, M.; Melara, G.; Sciorio, R.; Dallio, M.; Masarone, M.; Federico, A.; Persico, M. The role of resveratrol in liver disease: A comprehensive review from in vitro to clinical trials. Nutrients 2021, 13, 933.
- Nassir, F.; Ibdah, J.A. Sirtuins and nonalcoholic fatty liver disease. World J. Gastroenterol. 2016, 22, 10084–10092.
- Colak, Y.; Ozturk, O.; Senates, E.; Tuncer, I.; Yorulmaz, E.; Adali, G.; Doganay, L.; Enc, F.Y. SIRT1 as a potential therapeutic target for treatment of nonalcoholic fatty liver disease. Med. Sci. Monit. 2011, 17, 5–9.
- Ding, R.B.; Bao, J.L.; Deng, C.X. Emerging roles of SIRT1 in fatty liver diseases. Int. J. Biol. Sci. 2017, 13, 852–867.
- Hua, Y.Q.; Zeng, Y.; Xu, J.; Xu, X. Le Naringenin alleviates nonalcoholic steatohepatitis in middle-aged Apoe−/−mice: Role of SIRT1. Phytomedicine 2021, 81, 153412.
- Xu, X.; Zhu, X.P.; Bai, J.Y.; Xia, P.; Li, Y.; Lu, Y.; Li, X.Y.; Gao, X. Berberine alleviates nonalcoholic fatty liver induced by a high-fat diet in mice by activating SIRT3. FASEB J. 2019, 33, 7289–7300.
- de Gregorio, E.; Colell, A.; Morales, A.; Marí, M. Relevance of SIRT1-NF-κB axis as therapeutic target to ameliorate inflammation in liver disease. Int. J. Mol. Sci. 2020, 21, 3858.
- Ding, S.; Jiang, J.; Zhang, G.; Bu, Y.; Zhang, G.; Zhao, X. Resveratrol and caloric restriction prevent hepatic steatosis by regulating SIRT1-autophagy pathway and alleviating endoplasmic reticulum stress in high-fat diet-fed rats. PLoS ONE 2017, 12, e0183541.
- Li, C.X.; Gao, J.G.; Wan, X.Y.; Chen, Y.; Xu, C.F.; Feng, Z.M.; Zeng, H.; Lin, Y.M.; Ma, H.; Xu, P.; et al. Allyl isothiocyanate ameliorates lipid accumulation and inflammation in nonalcoholic fatty liver disease via the Sirt1/AMPK and NF-κB signaling pathways. World J. Gastroenterol. 2019, 25, 5120–5133.
- Rafiei, H.; Omidian, K.; Bandy, B. Dietary Polyphenols Protect Against Oleic Acid-Induced Steatosis in an in Vitro Model of NAFLD by Modulating Lipid Metabolism and Improving Mitochondrial Function. Nutrients 2019, 11, 541.
- Stacchiotti, A.; Grossi, I.; García-Gómez, R.; Patel, G.A.; Salvi, A.; Lavazza, A.; De Petro, G.; Monsalve, M.; Rezzani, R. Melatonin Effects on Non-Alcoholic Fatty Liver Disease Are Related to MicroRNA-34a-5p/Sirt1 Axis and Autophagy. Cells 2019, 8, 1053.
- Lee, Y.H.; Yun, M.R.; Kim, H.M.; Jeon, B.H.; Park, B.C.; Lee, B.W.; Kang, E.S.; Lee, H.C.; Park, Y.W.; Cha, B.S. Exogenous administration of DLK1 ameliorates hepatic steatosis and regulates gluconeogenesis via activation of AMPK. Int. J. Obes. 2016, 40, 356–365.
- Maharajan, N.; Ganesan, C.D.; Moon, C.; Jang, C.H.; Oh, W.K.; Cho, G.W. Licochalcone d ameliorates oxidative stress-induced senescence via ampk activation. Int. J. Mol. Sci. 2021, 22, 7324.
- Foretz, M.; Even, P.C.; Viollet, B. AMPK activation reduces hepatic lipid content by increasing fat oxidation in vivo. Int. J. Mol. Sci. 2018, 19, 2826.
- Salminen, A.; Kaarniranta, K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res. Rev. 2012, 11, 230–241.
- Han, X.; Tai, H.; Wang, X.; Wang, Z.; Zhou, J.; Wei, X.; Ding, Y.; Gong, H.; Mo, C.; Zhang, J.; et al. AMPK activation protects cells from oxidative stress-inducedsenescence via autophagic flux. Aging Cell 2016, 15, 416–427.
- Gao, Y.; Zhang, W.; Zeng, L.Q.; Bai, H.; Li, J.; Zhou, J.; Zhou, G.Y.; Fang, C.W.; Wang, F.; Qin, X.J. Exercise and dietary intervention ameliorate high-fat diet-induced NAFLD and liver aging by inducing lipophagy. Redox Biol. 2020, 36, 101635.
- Steinberg, G.R.; Carling, D. AMP-activated protein kinase: The current landscape for drug development. Nat. Rev. Drug Discov. 2019, 18, 527–551.
- de la Rosa, L.C.; Vrenken, T.E.; Buist-Homan, M.; Faber, K.N.; Moshage, H. Metformin protects primary rat hepatocytes against oxidative stress-induced apoptosis. Pharmacol. Res. Perspect. 2015, 3, e00125.
- Geng, Y.; Hernández Villanueva, A.; Oun, A.; Buist-Homan, M.; Blokzijl, H.; Faber, K.N.; Dolga, A.; Moshage, H. Protective effect of metformin against palmitate-induced hepatic cell death. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165621.
- Mora, F.A.A.; Musheshe, N.; Arroyave Ospina, J.C.; Geng, Y.; Soto, J.M.; Rodrigo, J.A.; Alieva, T.; Buist-Homan, M.; Lezoualc’h, F.; Cheng, X.; et al. Metformin protects against diclofenac-induced toxicity in primary rat hepatocytes by preserving mitochondrial integrity via a pathway involving EPAC. Biomed. Pharmacother. 2021, 143, 112072.
- Kulkarni, A.S.; Gubbi, S.; Barzilai, N. Benefits of Metformin in Attenuating the Hallmarks of Aging. Cell Metab. 2020, 32, 15–30.
- Le Pelletier, L.; Mantecon, M.; Gorwood, J.; Auclair, M.; Foresti, R.; Motterlini, R.; Laforge, M.; Atlan, M.; Fève, B.; Capeau, J.; et al. Metformin alleviates stress-induced cellular senescence of aging human adipose stromal cells and the ensuing adipocyte dysfunction. Elife 2021, 10, e62635.
- Kang, C. Senolytics and senostatics: A two-pronged approach to target cellular senescence for delaying aging and age-related diseases. Mol. Cells 2019, 42, 821–827.
- Mazzucco, A.E.; Smogorzewska, A.; Kang, C.; Luo, J.; Schlabach, M.R.; Xu, Q.; Patel, R.; Elledge, S.J. Genetic interrogation of replicative senescence uncovers a dual role for USP28 in coordinating the p53 and GATA4 branches of the senescence program. Genes Dev. 2017, 31, 1933–1938.
- De Cecco, M.; Ito, T.; Petrashen, A.P.; Elias, A.E.; Skvir, N.J.; Criscione, S.W.; Caligiana, A.; Brocculi, G.; Adney, E.M.; Boeke, J.D.; et al. LINE-1 derepression in senescent cells triggers interferon and inflammaging. Nature 2019, 566, 73.
- Zhang, Y.; Zhang, J.; Wang, S. The Role of Rapamycin in Healthspan Extension via the Delay of Organ Aging. Ageing Res. Rev. 2021, 70, 101376.
- Fok, W.C.; Chen, Y.; Bokov, A.; Zhang, Y.; Salmon, A.B.; Diaz, V.; Javors, M.; Wood, W.H., 3rd; Zhang, Y.; Becker, K.G.; et al. Mice Fed Rapamycin Have an Increase in Lifespan Associated with Major Changes in the Liver Transcriptome. PLoS ONE 2014, 9, e83988.
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016, 35, 724–742.
- Neef, M.; Ledermann, M.; Saegesser, H.; Schneider, V.; Reichen, J. Low-dose oral rapamycin treatment reduces fibrogenesis, improves liver function, and prolongs survival in rats with established liver cirrhosis. J. Hepatol. 2006, 45, 786–796.
- Martínez-Cisuelo, V.; Gómez, J.; García-Junceda, I.; Naudí, A.; Cabré, R.; Mota-Martorell, N.; López-Torres, M.; González-Sánchez, M.; Pamplona, R.; Barja, G. Rapamycin reverses age-related increases in mitochondrial ROS production at complex I, oxidative stress, accumulation of mtDNA fragments inside nuclear DNA, and lipofuscin level, and increases autophagy, in the liver of middle-aged mice. Exp. Gerontol. 2016, 83, 130–138.
- Cayo, A.; Segovia, R.; Venturini, W.; Moore-Carrasco, R.; Valenzuela, C.; Brown, N. Mtor activity and autophagy in senescent cells, a complex partnership. Int. J. Mol. Sci. 2021, 22, 8149.
- Moiseeva, O.; Deschênes-Simard, X.; St-Germain, E.; Igelmann, S.; Huot, G.; Cadar, A.E.; Bourdeau, V.; Pollak, M.N.; Ferbeyre, G. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-κB activation. Aging Cell 2013, 12, 489–498.
- Stavri, S.; Trusca, V.G.; Simionescu, M.; Gafencu, A.V. Metformin reduces the endotoxin-induced down-regulation of apolipoprotein e gene expression in macrophages. Biochem. Biophys. Res. Commun. 2015, 461, 435–440.
- Brandt, A.; Hernández-Arriaga, A.; Kehm, R.; Sánchez, V.; Jin, C.J.; Nier, A.; Baumann, A.; Camarinha-Silva, A.; Bergheim, I. Metformin attenuates the onset of non-alcoholic fatty liver disease and affects intestinal microbiota and barrier in small intestine. Sci. Rep. 2019, 9, 6668.
- Kita, Y.; Takamura, T.; Misu, H.; Ota, T.; Kurita, S.; Takeshita, Y.; Uno, M.; Matsuzawa-Nagata, N.; Kato, K.I.; Ando, H.; et al. Metformin Prevents and Reverses Inflammation in aNon-Diabetic Mouse Model of NonalcoholicSteatohepatitis.pdf. PLoS ONE 2012, 7, e43056.
- Garinis, G.A.; Fruci, B.; Mazza, A.; De Siena, M.; Abenavoli, S.; Gulletta, E.; Ventura, V.; Greco, M.; Abenavoli, L.; Belfiore, A. Metformin versus dietary treatment in nonalcoholic hepatic steatosis: A randomized study. Int. J. Obes. 2010, 34, 1255–1264.
- Laberge, R.; Zhou, L.; Sarantos, M.R.; Rodier, F.; De Keizer, P.L.J.; Liu, S.; Demaria, M.; Cong, Y.; Kapahi, P.; Desprez, P.; et al. Glucocorticoids Suppress Selected Components of the Senescence-Associated Secretory Phenotype. Aging Cell 2013, 11, 569–578.
- Miller, K.N.; Dasgupta, N.; Liu, T.; Adams, P.D.; Vizioli, M.G. Cytoplasmic chromatin fragments—from mechanisms to therapeutic potential. Elife 2021, 10, e63728.
- Wiley, C.D.; Schaum, N.; Alimirah, F.; Lopez-Dominguez, J.A.; Orjalo, A.V.; Scott, G.; Desprez, P.Y.; Benz, C.; Davalos, A.R.; Campisi, J. Small-molecule MDM2 antagonists attenuate the senescence-associated secretory phenotype. Sci. Rep. 2018, 8, 2–10.
- Xu, M.; Tchkonia, T.; Ding, H.; Ogrodnik, M.; Lubbers, E.R.; Pirtskhalava, T.; White, T.A.; Johnson, K.O.; Stout, M.B.; Mezera, V.; et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc. Natl. Acad. Sci. USA. 2015, 112, E6301–E6310.
More
Information
Subjects:
Gastroenterology & Hepatology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Revisions:
2 times
(View History)
Update Date:
06 Jun 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No