Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Johanna Arroyave-Ospina and Version 2 by Vivi Li.
Non-alcoholic fatty liver disease is characterized by disturbed lipid metabolism and increased oxidative stress. These conditions lead to the activation of different cellular response mechanisms, including senescence. Cellular senescence constitutes an important response to injury in the liver. Researcheent findings show that chronic oxidative stress can induce senescence, and this might be a driving mechanism for NAFLD progression, aggravating the disturbance of lipid metabolism, organelle dysfunction, pro-inflammatory response and hepatocellular damage. In this context, the modulation of cellular senescence can be beneficial to ameliorate oxidative stress-related damage during NAFLD progression.
- oxidative stress
- ROS
- non-alcoholic liver disease
- stress-induced senescence
- lipid metabolism
- ER stress
- mitochondrial dysfunction
1. Introduction
Non-alcoholic fatty liver disease (NAFLD) is a condition in which excessive amounts of lipids accumulate in the liver of individuals with no history of alcohol consumption, viral infection or (chronic) drug intoxication. NAFLD can progress to non-alcoholic steatohepatitis (NASH), a condition characterized by steatosis, inflammation, necrosis and fibrosis. The fibrotic stage can progress to cirrhosis and even hepatocellular carcinoma [1][2][1,2]. Among chronic liver diseases, NAFLD is considered the most prevalent worldwide, with its incidence and prevalence rapidly rising. The global prevalence of NAFLD is approximately 25%, with the highest prevalence reported in the Middle East (32%) and South America (31%), followed by Asia (27%), the USA (24%) and Europe (23%), whereas NAFLD is less common in Africa (14%) [3][4][3,4].
Oxidative stress (OxS) is an important factor in the pathogenesis of NAFLD [5]. OxS causes lipid and DNA damage, leading to organelle dysfunction, involving the impairment of mitochondrial and ER homeostasis, which may lead to senescence [6]. Senescence is a cellular program that induces prolonged or irreversible cell cycle arrest accompanied by distinct phenotypic alterations, including metabolic reprograming and chromatin remodeling. Senescent cells are characterized by at least two features: (1) resistance to apoptosis and (2) the acquisition of a complex pro-inflammatory secretome: the senescence-associated secretory phenotype or SASP [7]. Cellular senescence is triggered by different mechanisms and both endogenous and exogenous factors can induce premature senescence, e.g., lack of nutrients and/or growth factors, irreversible DNA damage, oncogene overexpression, mitochondrial dysfunction, oxidative stress and ER stress [8].
Several studies demonstrate that OxS is an important mediator of the initiation and progression of cellular senescence in the pathogenesis of chronic liver diseases, including NAFLD [9], and cellular senescence has been proposed to be a driving force of hepatic steatosis and an important contributing factor to NAFLD [10]. In this context, cellular senescence is activated as a cellular stress response to control liver damage and inflammation [11] and it has been demonstrated in both parenchymal as well as non-parenchymal liver cells [12].
2. Role of Senescence in the Development of NAFLD
2.1. Molecular Mechanisms of Cellular Senescence
Cellular senescence can be triggered by DNA damage caused by internal or external stimuli in chromosomes and telomeres. Two important mechanisms of cellular senescence are replicative senescence, mainly linked to telomere shortening during aging, and stress-induced senescence triggered by cellular stress, which is usually associated with DNA damage [13][29], also called premature senescence. Different stressors induce senescence, and they are often associated with p53 regulation. Some specific factors that determine stress-induced senescence are OxS, mitochondrial dysfunction and ER stress, which can induce irreversible DNA damage and activate the molecular mechanisms of senescence [14][30]. Stress factors induce DNA damage, in particular DNA double-strand breaks (DSBs), triggering the DNA damage response (DDR). The activation of this pathway leads to the activation of ATM (ataxia telangiectasia mutated) and Rad-3-related protein kinases, and subsequently to the phosphorylation of p53 and the activation of p21, which eventually results in cell cycle arrest. In addition, p21 and p16 inhibit the phosphorylation of the Rb protein (retinoblastoma factor), allowing it to bind to the E2F transcription factor and thus contributing to cell cycle arrest [14][30]. In summary, different cell signaling pathways related to cellular senescence converge in the activation of p53 and subsequently in the activation of the cyclin-dependent kinase (CDK) inhibitors p16 (INK4A), p15 (INK4B), p21 (WAF1) and p27 (CDKN1B) [15][31]. P53 levels determine cell fate during stress conditions and increased levels are observed in early senescence, but higher levels (at least two-times higher) are usually observed during apoptosis. Likewise, p53 levels can be stabilized and the maintenance of the senescence phenotype is often associated with p16 activation, when the senescent state is already stablished and it cannot be reversed by inhibiting p53 [16][32]. The DNA damage response is a well-known activator of SASP. The SASP includes an array of pro-inflammatory factors, mostly NF-kB dependent factors such as IL-6, IL-8, IL-1β, MCP-1 (monocyte chemoattractant protein-1), MCP-2 and MCP-4, growth factors such as HGF (human growth factor) and FGF (fibroblast growth factor), proteases such as MMPs (matrix metalloproteinases) and secreted insoluble proteins/extracellular matrix proteins (ECM). SASP can reinforce senescence-induced cell cycle arrest by autocrine or paracrine mechanisms and can modulate the tissue microenvironment by paracrine pathways [17][33]. The SASP is induced and regulated by several signaling pathways, leading to the activation of NF-κB and/or CCAAT/enhancer-binding protein-β (C/EBPβ). These pathways include the NOTCH signaling pathway [18][34], the cGAS–STING pathway [19][20][35,36] and the NAD+–NAMPT pathway [21][37]. Furthermore, the SASP seems to be context dependent and its composition varies depending on the type of stimulus, duration and cell type [14][30].2.2. Senescence Features in Liver Disease
Hepatocyte senescence can be induced by various stress conditions, such as metabolic stress, viral infections and oncogenic stimuli and it can also be induced by aging [22][38]. The senescence of hepatocytes appears to be a hallmark in chronic liver disease independent of the etiology. The normal liver contains a rather constant level of senescent hepatocytes (3–7%), but in chronic liver diseases, this percentage may increase to 50–100% [23][39]. Chronic liver injury contributes to the continuous generation of senescent cells or “chronic” senescence, aggravating liver dysfunction and tumor progression [9]. Moreover, chronic liver injury induces functional changes in hepatocytes, which, sometimes, can be reversed or progress to cell death, depending on the level of injury [22][38]. In general, senescent hepatocytes undergo morphological and biochemical changes similar to those already reported as markers of cellular senescence, including a reduced number of mitochondria (decreased biogenesis and/or increased mitophagy) [24][40], the accumulation of lipofuscin, the increased expression and/or activity of HP1β, p21, p16, p53 and γ-H2AX and elevated SA-β-galactosidase activity [25][26][27][41,42,43]. Hepatocyte senescence also leads to the SASP [28][44], resulting in the production of pro-inflammatory mediators and alterations in the tissue microenvironment. For instance, TGF-β signaling appears to be involved in the “spreading” of senescence from one cell to another. When TGF-β signaling is inhibited in mice, senescence is impeded, regeneration is accelerated and survival is improved [29][45]. It has also been shown that IL-1 and IL-6 are capable of inducing senescence in adjacent cells, which is antagonized by the inhibition of their receptors [23][39]. During liver injury of various etiology, including NAFLD, damage-associated molecular patterns (DAMPs) link inflammation to cell death mechanisms [26][42]. Likewise, cellular senescence is activated as an endogenous response mechanism in cellular stress conditions. Senescence of various parenchymal and non-parenchymal resident liver cells has been demonstrated in chronic liver diseases [9]. Inflammation plays a crucial role in the development of liver injury due the involvement of circulating and resident immune cells e.g., neutrophils, monocytes and macrophages, together with liver non-parenchymal cells such as Kupffer cells (KCs), liver sinusoidal endothelial cells (LSECs) and hepatic stellate cells (HSCs). Progression of NAFLD is often accompanied by inflammatory events and clinical studies have shown that progression of the disease is often associated with excessive activation of the innate and the adaptive immune response [30][46]. Lipotoxicity and organelle dysfunction, mediated by OxS, induce hepatocyte cell death, which is a key trigger of liver inflammation in NAFLD. In addition, KCs are activated by phagocytosis of apoptotic bodies leading to the production of TNF, TRAIL and FAS ligand further promoting hepatocyte apoptosis, liver inflammation and fibrosis [31][47]. Characterization of antigenic stimuli triggering the adaptive immune response in the liver will also be important to elucidate the role of the adaptive immune system in liver inflammation and damage. As already mentioned, OxS and lipid peroxidation are common features of NAFLD. Oxidized phospholipids and reactive aldehydes produced during lipid peroxidation, such as malondialdehyde, induce hepatic inflammation, but also form antigenic adducts with cellular macromolecules known as OxS-derived epitopes (OSEs) [32][48]. The involvement of OxS in driving NAFLD-associated immune responses was inferred from observations that elevated titers of anti-OSE IgG were detected in approximately 40% of patients with NAFLD or NASH in two unrelated cohorts [33][49]. These studies demonstrate that OxS is an important contributing factor in the immune response during progression of NAFLD. Likewise, high titers of anti-OSE IgGs are associated with the severity of lobular inflammation, the prevalence of intrahepatic B cell and/or T cell aggregates and are an independent predictor of fibrosis [32][34][48,50]. Moreover, in liver injury conditions neutrophils promote ROS production via enzymatic activation of NADPH oxidase, activation of proteases and synthesis of inflammatory mediators that serve as chemo-attractants of other immune cells such as macrophages [35][36][51,52]. KCs are directly involved in the progression of NAFLD, since they contribute importantly to hepatic ROS production and release of fibrogenic cytokines such as TGF-β [37][53], promoting the activation of hepatic stellate cells via SMAD signaling [38][54]. Considering the participation of immune cells in NAFLD development, it is relevant to mention that neutrophils can trigger telomere dysfunction via ROS production in an in vivo model of acute liver injury using carbon tetrachloride (CCl4). Besides, it has been found that neutrophils can reduce in vitro cell growth of co-cultured fibroblasts, with elevation of senescence markers and high ROS production [39][55], suggesting that senescence mediated by inflammatory cells is (partly) ROS dependent. In addition, senescent cells mediate the recruitment of neutrophils, thereby aggravating ROS production and inflammation [39][55]. Interestingly, inflammatory cells are a hallmark of steatohepatitis in NAFLD and increased ROS production can influence directly and indirectly the activation of liver inflammatory cells, a process that might be mediated by the SASP enhancing liver damage and promoting fibrogenesis during NAFLD.3. Molecular Mechanisms of Oxidative Stress Contributing to Senescence in NAFLD
3.1. Senescence and Oxidative Stress
The role of OxS in the modulation of senescence and excessive ROS production has been identified as a driving factor of senescence [6]. It has been observed that the induction of ROS production in vitro recapitulated senescence-induced changes in healthy at term placental explants. This was demonstrated to be tightly dependent on p21 expression, which might be regulated by OxS [40][41][42][78,79,80]. Cellular senescence also correlates with OxS-induced damage in various tissues and/or cell populations. For instance, increased levels of 8-hydroxy-2′-deoxyguanosine (8-OHdG), a marker of DNA oxidative damage and ROS production have been found to be associated with senescence in different cell types [43][44][45][81,82,83]. Moreover, the induction of OxS is enough to induce cell cycle arrest, as also demonstrated after exposure of IMR-90 cells to hydrogen peroxide (H2O2), in which long-term growth arrest, morphological changes and the activation of SA-β-galactosidase with increased p21 expression were observed [42][80]. Experiments with endothelial cells have also demonstrated that ROS production induced by hydrogen peroxide treatment correlates with the induction of senescence [43][81]. NAFLD development and progression are closely related to OxS and increasing experimental evidence suggests that OxS is a critical factor in the regulation of cellular senescence in NAFLD [44][82]. In the next section, reswearchers discuss the mechanisms of OxS-induced senescence in hepatocytes and its role in the progression of NAFLD. The main mechanisms and features of OxS-induced senescence in NAFLD are summarized in Figure 12.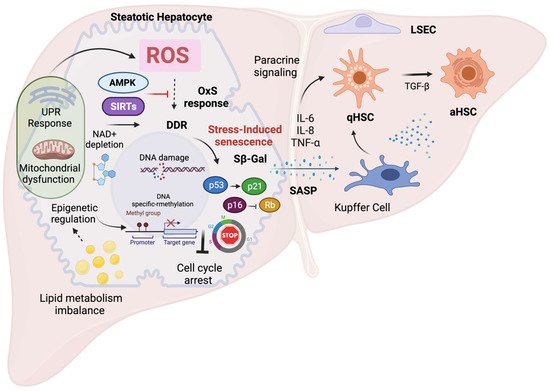
Figure 12. Mechanisms and features of oxidative stress-induced senescenceExcess ROS triggers premature senescence as part of the cellular stress response via activation of the DNA-damage response with concomitant activation of p53-p21 and p16-Rb pathways. This causes prolonged cell cycle arrest and prevents the activation of the cell death program and limits liver damage. Impaired lipid metabolism during NAFLD induces organelle dysfunction, contributing to OxS and senescence. Chronic oxidative stress during NAFLD leads to dysregulation of several factors such as depletion of NAD+ levels with diminished SIRT expression and downregulation of AMPK signaling, leading to deleterious cellular senescence. These pathways can be potential therapeutic targets to control cellular senescence via modulation of OxS. Other cellular response mechanisms such as epigenetic regulation, can directly influence cellular senescence, e.g., via p16 activation. Epigenetic changes have been demonstrated to be induced by lipid accumulation and OxS during NAFLD. Finally, the SASP along with the production of pro-inflammatory factors can influence neighboring cells by paracrine signaling (bystander effect), and allows activation of non-parenchymal cells contributing to NAFLD progression.