 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Monica Embers | -- | 3500 | 2022-06-01 18:37:28 | | | |
| 2 | Catherine Yang | Meta information modification | 3500 | 2022-06-06 04:48:54 | | |
Video Upload Options
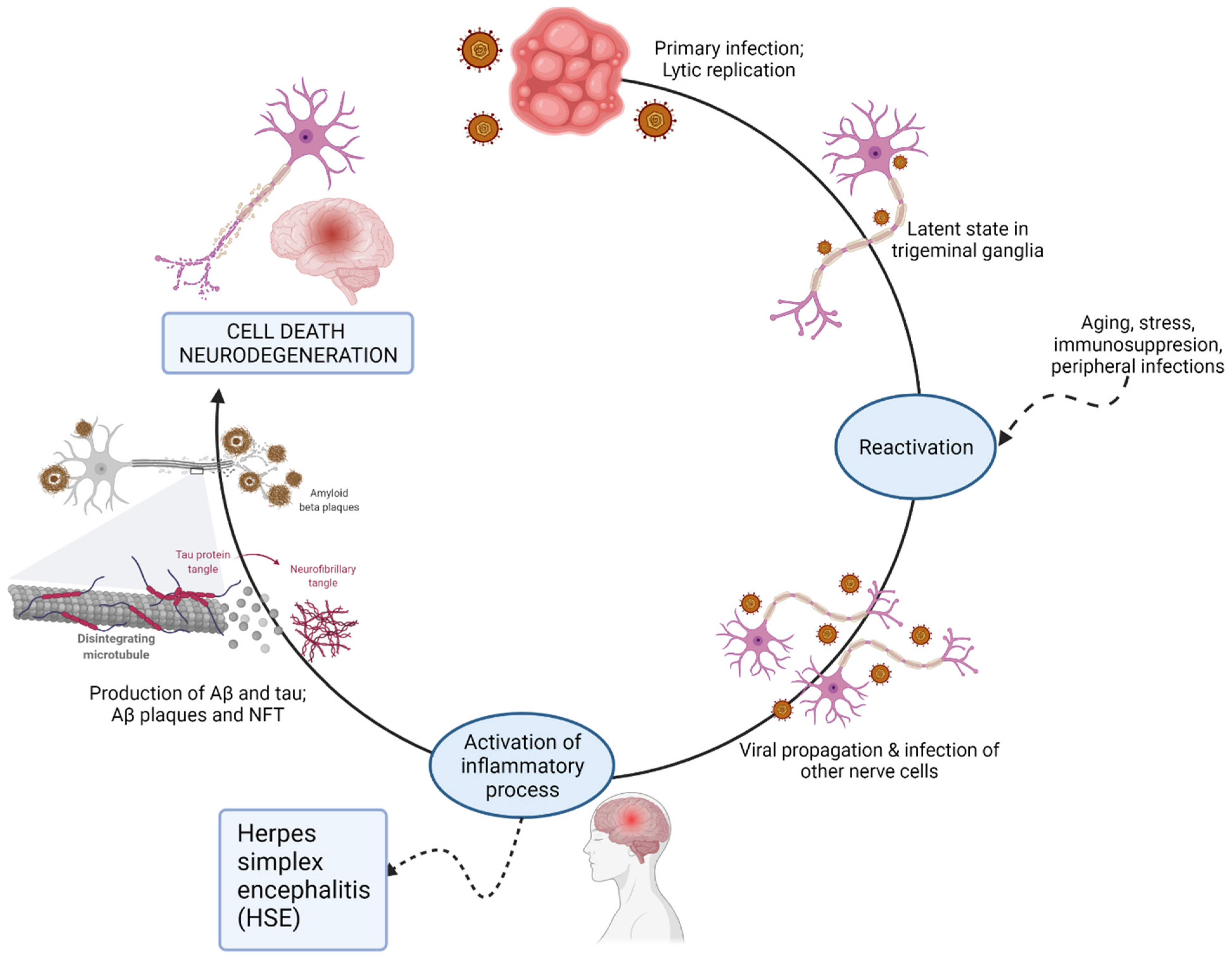
The potential contribution of pathogenic microbes to dementia-inducing disease is a subject of considerable importance. Alzheimer’s disease (AD) is a neurocognitive disease that slowly destroys brain function, leading to cognitive decline and behavioral and psychiatric disorders. The histopathology of AD is associated with neuronal loss and progressive synaptic dysfunction, accompanied by the deposition of amyloid-β (Aβ) peptide in the form of parenchymal plaques and abnormal aggregated tau protein in the form of neurofibrillary tangles. The AD pathogen hypothesis states that pathogens and microbes act as triggers, interacting with genetic factors to initiate the accumulation of Aβ, hyperphosphorylated tau protein (p-tau), and inflammation in the brain. Evidence indicates that Borrelia sp., HSV-1, VZV (HHV-2), HHV-6/7, oral pathogens, Chlamydophila pneumoniae, and Candida albicans can infect the central nervous system (CNS), evade the immune system, and consequently prevail in the AD brain.
1. The Herpesviridae Family
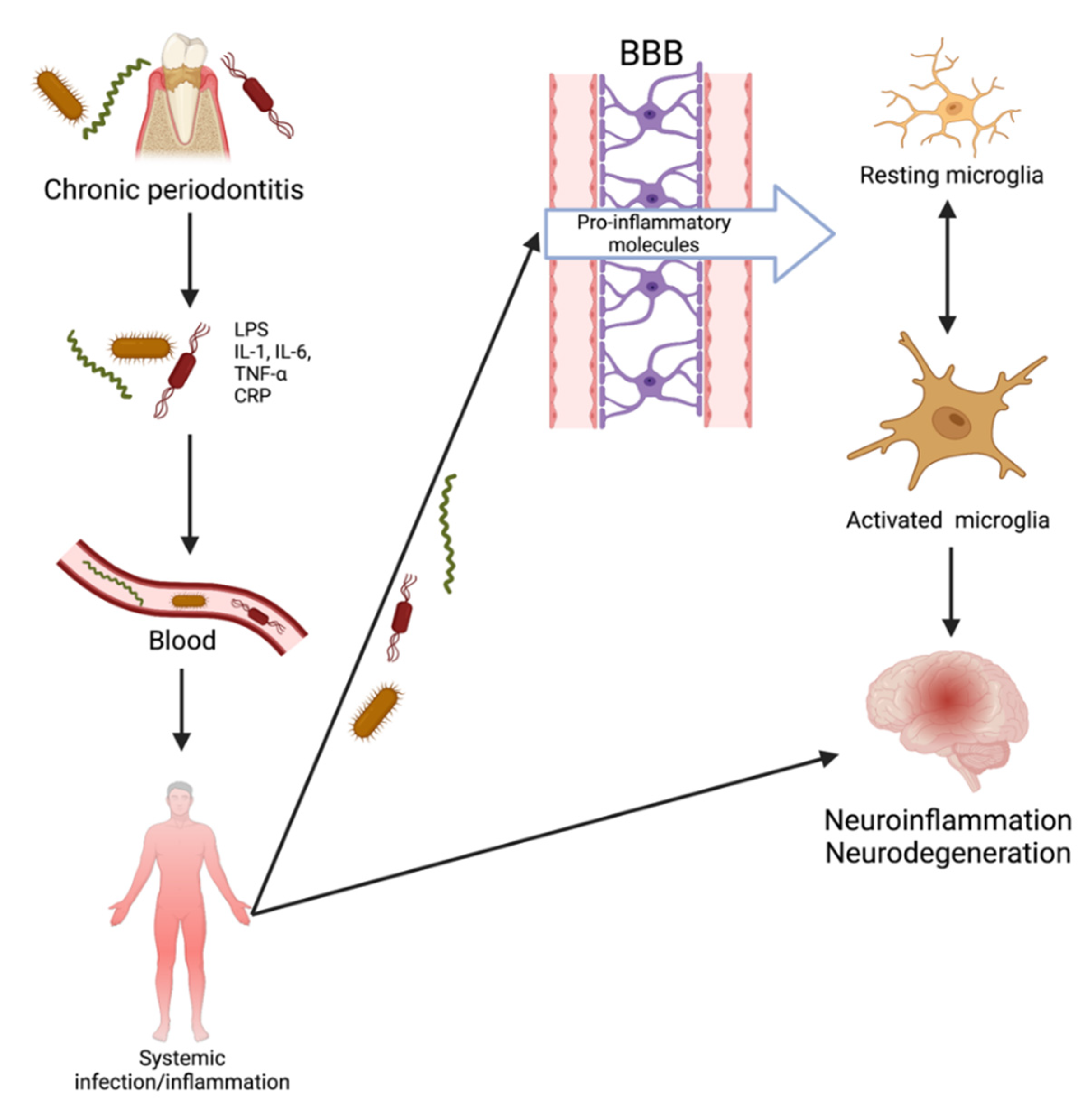
2. Oral Bacteria
3. Borrelia burgdorferi
4. Chlamydia pneumoniae
5. Fungal Pathogens
References
- Itzhaki, R.F. Corroboration of a Major Role for Herpes Simplex Virus Type 1 in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 324.
- McQuillan, G.; Kruszon-Moran, D.; Flagg, E.W.; Paulose-Ram, R. Prevalence of Herpes Simplex Virus Type 1 and Type 2 in Persons Aged 14–49: United States, 2015–2016; National Center for Health Statistics: Hyattsville, MD, USA, 2018; pp. 1–8.
- Suzich, J.B.; Cliffe, A.R. Strength in diversity: Understanding the pathways to herpes simplex virus reactivation. Virology 2018, 522, 81–91.
- Ball, M.J. Limbic predilection in Alzheimer dementia: Is reactivated herpesvirus involved? Can. J. Neurol. Sci. 1982, 9, 303–306.
- Harris, S.A.; Harris, E.A. Herpes Simplex Virus Type 1 and Other Pathogens are Key Causative Factors in Sporadic Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 48, 319–353.
- Liang, Y.; Zhou, Z.; Wang, H.; Cheng, X.; Zhong, S.; Zhao, C. Association of apolipoprotein E genotypes with epilepsy risk: A systematic review and meta-analysis. Epilepsy Behav. 2019, 98, 27–35.
- Lamoureux, L.; Marottoli, F.M.; Tseng, K.Y.; Tai, L.M. APOE4 Promotes Tonic-Clonic Seizures, an Effect Modified by Familial Alzheimer’s Disease Mutations. Front. Cell Dev. Biol. 2021, 9, 656521.
- Tzeng, N.S.; Chung, C.H.; Lin, F.H.; Chiang, C.P.; Yeh, C.B.; Huang, S.Y.; Lu, R.B.; Chang, H.A.; Kao, Y.C.; Yeh, H.W.; et al. Anti-herpetic Medications and Reduced Risk of Dementia in Patients with Herpes Simplex Virus Infections—A Nationwide, Population-Based Cohort Study in Taiwan. Neurotherapeutics 2018, 15, 417–429.
- Jamieson, G.A.; Maitland, N.J.; Craske, J.; Wilcock, G.K.; Itzhaki, R.F. Detection of herpes simplex virus type 1 DNA sequences in normal and Alzheimer’s disease brain using polymerase chain reaction. Biochem. Soc. Trans. 1991, 19, 122S.
- Itzhaki, R.F. Herpes simplex virus type 1 and Alzheimer’s disease: Possible mechanisms and signposts. FASEB J. 2017, 31, 3216–3226.
- Licastro, F.; Carbone, I.; Raschi, E.; Porcellini, E. The 21st century epidemic: Infections as inductors of neuro-degeneration associated with Alzheimer’s Disease. Immun. Ageing 2014, 11, 22.
- Readhead, B.; Haure-Mirande, J.V.; Funk, C.C.; Richards, M.A.; Shannon, P.; Haroutunian, V.; Sano, M.; Liang, W.S.; Beckmann, N.D.; Price, N.D.; et al. Multiscale Analysis of Independent Alzheimer’s Cohorts Finds Disruption of Molecular, Genetic, and Clinical Networks by Human Herpesvirus. Neuron 2018, 99, 64–82.e67.
- Komaroff, A.L.; Boeckh, M.; Eliason, E.; Phan, T.; Kaufer, B.B. Summary of the 10th International Conference on Human Herpesviruses-6 and -7 (HHV-6A, -6B, and HHV-7). J. Med. Virol. 2018, 90, 625–630.
- Wang, Y.; Ding, L.; Zhu, Q.; Shu, M.; Cai, Q. Common Infections May Lead to Alzheimer’s Disease. Virol. Sin. 2018, 33, 456–458.
- Fenno, J.C. Treponema denticola interactions with host proteins. J. Oral. Microbiol. 2012, 4, 9929.
- Mira, A.; Simon-Soro, A.; Curtis, M.A. Role of microbial communities in the pathogenesis of periodontal diseases and caries. J. Clin. Periodontol. 2017, 44 (Suppl. 18), S23–S38.
- Kamer, A.R.; Dasanayake, A.P.; Craig, R.G.; Glodzik-Sobanska, L.; Bry, M.; de Leon, M.J. Alzheimer’s disease and peripheral infections: The possible contribution from periodontal infections, model and hypothesis. J. Alzheimer’s Dis. 2008, 13, 437–449.
- Abbayya, K.; Puthanakar, N.Y.; Naduwinmani, S.; Chidambar, Y.S. Association between Periodontitis and Alzheimer’s Disease. N. Am. J. Med. Sci. 2015, 7, 241–246.
- Hasegawa-Ishii, S.; Shimada, A.; Imamura, F. Lipopolysaccharide-initiated persistent rhinitis causes gliosis and synaptic loss in the olfactory bulb. Sci. Rep. 2017, 7, 11605.
- Leira, Y.; Iglesias-Rey, R.; Gomez-Lado, N.; Aguiar, P.; Campos, F.; D’Aiuto, F.; Castillo, J.; Blanco, J.; Sobrino, T. Porphyromonas gingivalis lipopolysaccharide-induced periodontitis and serum amyloid-beta peptides. Arch. Oral Biol. 2019, 99, 120–125.
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5, eaau3333.
- Ilievski, V.; Zuchowska, P.K.; Green, S.J.; Toth, P.T.; Ragozzino, M.E.; Le, K.; Aljewari, H.W.; O’Brien-Simpson, N.M.; Reynolds, E.C.; Watanabe, K. Chronic oral application of a periodontal pathogen results in brain inflammation, neurodegeneration and amyloid beta production in wild type mice. PLoS ONE 2018, 13, e0204941.
- Ding, Y.; Ren, J.; Yu, H.; Yu, W.; Zhou, Y. Porphyromonas gingivalis, a periodontitis causing bacterium, induces memory impairment and age-dependent neuroinflammation in mice. Immun. Ageing 2018, 15, 6.
- Zhang, J.; Yu, C.; Zhang, X.; Chen, H.; Dong, J.; Lu, W.; Song, Z.; Zhou, W. Porphyromonas gingivalis lipopolysaccharide induces cognitive dysfunction, mediated by neuronal inflammation via activation of the TLR4 signaling pathway in C57BL/6 mice. J. Neuroinflamm. 2018, 15, 37.
- Singhrao, S.K.; Chukkapalli, S.; Poole, S.; Velsko, I.; Crean, S.J.; Kesavalu, L. Chronic Porphyromonas gingivalis infection accelerates the occurrence of age-related granules in ApoE(−)(/)(−) mice brains. J. Oral Microbiol. 2017, 9, 1270602.
- Wu, Z.; Ni, J.; Liu, Y.; Teeling, J.L.; Takayama, F.; Collcutt, A.; Ibbett, P.; Nakanishi, H. Cathepsin B plays a critical role in inducing Alzheimer’s disease-like phenotypes following chronic systemic exposure to lipopolysaccharide from Porphyromonas gingivalis in mice. Brain Behav. Immun. 2017, 65, 350–361.
- Liu, Y.; Wu, Z.; Nakanishi, Y.; Ni, J.; Hayashi, Y.; Takayama, F.; Zhou, Y.; Kadowaki, T.; Nakanishi, H. Infection of microglia with Porphyromonas gingivalis promotes cell migration and an inflammatory response through the gingipain-mediated activation of protease-activated receptor-2 in mice. Sci. Rep. 2017, 7, 11759.
- Ishida, N.; Ishihara, Y.; Ishida, K.; Tada, H.; Funaki-Kato, Y.; Hagiwara, M.; Ferdous, T.; Abdullah, M.; Mitani, A.; Michikawa, M.; et al. Periodontitis induced by bacterial infection exacerbates features of Alzheimer’s disease in transgenic mice. NPJ Aging Mech. Dis. 2017, 3, 15.
- Poole, S.; Singhrao, S.K.; Chukkapalli, S.; Rivera, M.; Velsko, I.; Kesavalu, L.; Crean, S. Active invasion of Porphyromonas gingivalis and infection-induced complement activation in ApoE−/− mice brains. J. Alzheimer’s Dis. 2015, 43, 67–80.
- Miklossy, J. Bacterial Amyloid and DNA are Important Constituents of Senile Plaques: Further Evidence of the Spirochetal and Biofilm Nature of Senile Plaques. J. Alzheimer’s Dis. 2016, 53, 1459–1473.
- Miklossy, J. Historic evidence to support a causal relationship between spirochetal infections and Alzheimer’s disease. Front. Aging Neurosci. 2015, 7, 46.
- Pacheco e Silva, A.C. Espirochetose dos centros nervos. Mem. Hospicio Juquery 1926, 3–4, 1–27.
- Miklossy, J. Alzheimer’s disease—A spriochetosis? Neuroreport 1993, 4, 841–848.
- Kugeler, K.J.; Schwartz, A.M.; Delorey, M.J.; Mead, P.S.; Hinckley, A.F. Estimating the Frequency of Lyme Disease Diagnoses, United States, 2010–2018. Emerg. Infect. Dis. 2021, 27, 616–619.
- Schwartz, A.M.; Kugeler, K.J.; Nelson, C.A.; Marx, G.E.; Hinckley, A.F. Use of Commercial Claims Data for Evaluating Trends in Lyme Disease Diagnoses, United States, 2010–2018. Emerg. Infect. Dis. 2021, 27, 499–507.
- Ramesh, G.; Borda, J.T.; Dufour, J.; Kaushal, D.; Ramamoorthy, R.; Lackner, A.A.; Philipp, M.T. Interaction of the Lyme disease spirochete Borrelia burgdorferi with brain parenchyma elicits inflammatory mediators from glial cells as well as glial and neuronal apoptosis. Am. J. Pathol. 2008, 173, 1415–1427.
- Luft, B.J.; Steinman, C.R.; Neimark, H.C.; Muralidhar, B.; Rush, T.; Finkel, M.F.; Kunkel, M.; Dattwyler, R.J. Invasion of the central nervous system by Borrelia burgdorferi in acute disseminated infection. JAMA 1992, 267, 1364–1367.
- Maksimyan, S.; Syed, M.S.; Soti, V. Post-Treatment Lyme Disease Syndrome: Need for Diagnosis and Treatment. Cureus 2021, 13, e18703.
- Crossland, N.A.; Alvarez, X.; Embers, M.E. Late Disseminated Lyme Disease: Associated Pathology and Spirochete Persistence Posttreatment in Rhesus Macaques. Am. J. Pathol. 2018, 188, 672–682.
- Embers, M.E.; Barthold, S.W.; Borda, J.T.; Bowers, L.; Doyle, L.; Hodzic, E.; Jacobs, M.B.; Hasenkampf, N.R.; Martin, D.S.; Narasimhan, S.; et al. Persistence of Borrelia burgdorferi in rhesus macaques following antibiotic treatment of disseminated infection. PLoS ONE 2012, 7, e29914.
- Hodzic, E.; Imai, D.; Feng, S.; Barthold, S.W. Resurgence of persisting non-cultivable Borrelia burgdorferi following antibiotic treatment in mice. PLoS ONE 2014, 9, e86907.
- Garcia-Monco, J.C.; Villar, B.F.; Alen, J.C.; Benach, J.L. Borrelia burgdorferi in the central nervous system: Experimental and clinical evidence for early invasion. J. Infect. Dis. 1990, 161, 1187–1193.
- MacDonald, A.B.; Miranda, J.M. Concurrent neocortical borreliosis and Alzheimer’s disease. Hum. Pathol. 1987, 18, 759–761.
- Meer-Scherrer, L.; Chang Loa, C.; Adelson, M.E.; Mordechai, E.; Lobrinus, J.A.; Fallon, B.A.; Tilton, R.C. Lyme disease associated with Alzheimer’s disease. Curr. Microbiol. 2006, 52, 330–332.
- Miklossy, J.; Khalili, K.; Gern, L.; Ericson, R.L.; Darekar, P.; Bolle, L.; Hurlimann, J.; Paster, B.J. Borrelia burgdorferi persists in the brain in chronic lyme neuroborreliosis and may be associated with Alzheimer disease. J. Alzheimer’s Dis. 2004, 6, 639–649.
- Miklossy, J. Alzheimer’s disease—A neurospirochetosis. Analysis of the evidence following Koch’s and Hill’s criteria. J. Neuroinflamm. 2011, 8, 90.
- Pappolla, M.A.; Omar, R.; Saran, B.; Andorn, A.; Suarez, M.; Pavia, C.; Weinstein, A.; Shank, D.; Davis, K.; Burgdorfer, W. Concurrent neuroborreliosis and Alzheimer’s disease: Analysis of the evidence. Hum. Pathol. 1989, 20, 753–757.
- Gutacker, M.; Valsangiacomo, C.; Balmelli, T.; Bernasconi, M.V.; Bouras, C.; Piffaretti, J.C. Arguments against the involvement of Borrelia burgdorferi sensu lato in Alzheimer’s disease. Res. Microbiol. 1998, 149, 31–37.
- Marques, A.R.; Weir, S.C.; Fahle, G.A.; Fischer, S.H. Lack of evidence of Borrelia involvement in Alzheimer’s disease. J. Infect. Dis. 2000, 182, 1006–1007.
- McLaughlin, R.; Kin, N.M.; Chen, M.F.; Nair, N.P.; Chan, E.C. Alzheimer’s disease may not be a spirochetosis. Neuroreport 1999, 1, 1489–1491.
- Galbussera, A.; Tremolizzo, L.; Isella, V.; Gelosa, G.; Vezzo, R.; Vigorè, L.; Brenna, M.; Ferrarese, C.; Appollonio, I. Lack of evidence for Borrelia burgdorferi seropositivity in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2008, 22, 308.
- Kugeler, K.J.; Farley, G.M.; Forrester, J.D.; Mead, P.S. Geographic Distribution and Expansion of Human Lyme Disease, United States. Emerg. Infect. Dis. 2015, 21, 1455–1457.
- Schumacher, H.R., Jr.; Gerard, H.C.; Arayssi, T.K.; Pando, J.A.; Branigan, P.J.; Saaibi, D.L.; Hudson, A.P. Lower prevalence of Chlamydia pneumoniae DNA compared with Chlamydia trachomatis DNA in synovial tissue of arthritis patients. Arthritis Rheum. 1999, 42, 1889–1893.
- Belland, R.J.; Ouellette, S.P.; Gieffers, J.; Byrne, G.I. Chlamydia pneumoniae and atherosclerosis. Cell Microbiol. 2004, 6, 117–127.
- Wagner, A.D.; Gerard, H.C.; Fresemann, T.; Schmidt, W.A.; Gromnica-Ihle, E.; Hudson, A.P.; Zeidler, H. Detection of Chlamydia pneumoniae in giant cell vasculitis and correlation with the topographic arrangement of tissue-infiltrating dendritic cells. Arthritis Rheum. 2000, 43, 1543–1551.
- Shima, K.; Kuhlenbaumer, G.; Rupp, J. Chlamydia pneumoniae infection and Alzheimer’s disease: A connection to remember? Med. Microbiol. Immunol. 2010, 199, 283–289.
- Little, C.S.; Joyce, T.A.; Hammond, C.J.; Matta, H.; Cahn, D.; Appelt, D.M.; Balin, B.J. Detection of bacterial antigens and Alzheimer’s disease-like pathology in the central nervous system of BALB/c mice following intranasal infection with a laboratory isolate of Chlamydia pneumoniae. Front. Aging Neurosci. 2014, 6, 304.
- Balin, B.J.; Gérard, H.C.; Arking, E.J.; Appelt, D.M.; Branigan, P.J.; Abrams, J.T.; Whittum-Hudson, J.A.; Hudson, A.P. Identification and localization of Chlamydia pneumoniae in the Alzheimer’s brain. Med. Microbiol. Immunol. 1998, 187, 23–42.
- Gerard, H.C.; Dreses-Werringloer, U.; Wildt, K.S.; Deka, S.; Oszust, C.; Balin, B.J.; Frey, W.H., 2nd; Bordayo, E.Z.; Whittum-Hudson, J.A.; Hudson, A.P. Chlamydophila (Chlamydia) pneumoniae in the Alzheimer’s brain. FEMS Immunol. Med. Microbiol. 2006, 48, 355–366.
- Appelt, D.M.; Roupas, M.R.; Way, D.S.; Bell, M.G.; Albert, E.V.; Hammond, C.J.; Balin, B.J. Inhibition of apoptosis in neuronal cells infected with Chlamydophila (Chlamydia) pneumoniae. BMC Neurosci. 2008, 9, 13.
- Hammond, C.J.; Hallock, L.R.; Howanski, R.J.; Appelt, D.M.; Little, C.S.; Balin, B.J. Immunohistological detection of Chlamydia pneumoniae in the Alzheimer’s disease brain. BMC Neurosci. 2010, 11, 121.
- Boelen, E.; Steinbusch, H.W.; van der Ven, A.J.; Grauls, G.; Bruggeman, C.A.; Stassen, F.R. Chlamydia pneumoniae infection of brain cells: An in vitro study. Neurobiol. Aging 2007, 28, 524–532.
- MacIntyre, A.; Hammond, C.J.; Little, C.S.; Appelt, D.M.; Balin, B.J. Chlamydia pneumoniae infection alters the junctional complex proteins of human brain microvascular endothelial cells. FEMS Microbiol. Lett. 2002, 217, 167–172.
- MacIntyre, A.; Abramov, R.; Hammond, C.J.; Hudson, A.P.; Arking, E.J.; Little, C.S.; Appelt, D.M.; Balin, B.J. Chlamydia pneumoniae infection promotes the transmigration of monocytes through human brain endothelial cells. J. Neurosci. Res. 2003, 71, 740–750.
- Sochocka, M.; Zwolinska, K.; Leszek, J. The Infectious Etiology of Alzheimer’s Disease. Curr. Neuropharmacol. 2017, 15, 996–1009.
- Dreses-Werringloer, U.; Gerard, H.C.; Whittum-Hudson, J.A.; Hudson, A.P. Chlamydophila (Chlamydia) pneumoniae infection of human astrocytes and microglia in culture displays an active, rather than a persistent, phenotype. Am. J. Med. Sci. 2006, 332, 168–174.
- Dreses-Werringloer, U.; Bhuiyan, M.; Zhao, Y.; Gerard, H.C.; Whittum-Hudson, J.A.; Hudson, A.P. Initial characterization of Chlamydophila (Chlamydia) pneumoniae cultured from the late-onset Alzheimer brain. Int. J. Med. Microbiol. 2009, 299, 187–201.
- Arking, E.J.; Appelt, D.M.; Abrams, J.T.; Kolbe, S.; Hudson, A.P.; Balin, B.J. Ultrastructural Analysis of Chlamydia pneumoniae in the Alzheimer’s Brain. Pathogenesis 1999, 1, 201–211.
- Balin, B.J.; Hammond, C.J.; Little, C.S.; Hingley, S.T.; Al-Atrache, Z.; Appelt, D.M.; Whittum-Hudson, J.A.; Hudson, A.P. Chlamydia pneumoniae: An Etiologic Agent for Late-Onset Dementia. Front. Aging Neurosci. 2018, 10, 302.
- Paradowski, B.; Jaremko, M.; Dobosz, T.; Leszek, J.; Noga, L. Evaluation of CSF-Chlamydia pneumoniae, CSF-tau, and CSF-Abeta42 in Alzheimer’s disease and vascular dementia. J. Neurol. 2007, 254, 154–159.
- Nochlin, D.; Shaw, C.M.; Campbell, L.A.; Kuo, C.C. Failure to detect Chlamydia pneumoniae in brain tissues of Alzheimer’s disease. Neurology 1999, 53, 1888.
- Gieffers, J.; Reusche, E.; Solbach, W.; Maass, M. Failure to detect Chlamydia pneumoniae in brain sections of Alzheimer’s disease patients. J. Clin. Microbiol. 2000, 38, 881–882.
- Tsay, S.; Williams, S.; Mu, Y.; Epson, E.; Johnston, H.; Farley, M.M.; Harrison, L.H.; Vonbank, B.; Shrum, S.; Dumyati, G.; et al. National Burden of Candidemia, United States, 2017. Open Forum Infect. Dis. 2018, 5, S142–S143.
- Parady, B. Innate Immune and Fungal Model of Alzheimer’s Disease. J. Alzheimer’s Dis. Rep. 2018, 2, 139–152.
- Pisa, D.; Alonso, R.; Rabano, A.; Rodal, I.; Carrasco, L. Different Brain Regions are Infected with Fungi in Alzheimer’s Disease. Sci. Rep. 2015, 5, 15015.
- Pisa, D.; Alonso, R.; Rabano, A.; Carrasco, L. Corpora Amylacea of Brain Tissue from Neurodegenerative Diseases Are Stained with Specific Antifungal Antibodies. Front. Neurosci. 2016, 10, 86.
- Pisa, D.; Alonso, R.; Juarranz, A.; Rabano, A.; Carrasco, L. Direct visualization of fungal infection in brains from patients with Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 43, 613–624.
- Alonso, R.; Pisa, D.; Rabano, A.; Carrasco, L. Alzheimer’s disease and disseminated mycoses. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 1125–1132.
- Pisa, D.; Alonso, R.; Rabano, A.; Horst, M.N.; Carrasco, L. Fungal Enolase, beta-Tubulin, and Chitin Are Detected in Brain Tissue from Alzheimer’s Disease Patients. Front. Microbiol. 2016, 7, 1772.
- Roberts, R.O.; Christianson, T.J.; Kremers, W.K.; Mielke, M.M.; Machulda, M.M.; Vassilaki, M.; Alhurani, R.E.; Geda, Y.E.; Knopman, D.S.; Petersen, R.C. Association Between Olfactory Dysfunction and Amnestic Mild Cognitive Impairment and Alzheimer Disease Dementia. JAMA Neurol. 2016, 73, 93–101.
- Hernandez-Chavez, M.J.; Perez-Garcia, L.A.; Nino-Vega, G.A.; Mora-Montes, H.M. Fungal Strategies to Evade the Host Immune Recognition. J. Fungi 2017, 3, 51.
- Höfs, S.; Mogavero, S.; Hube, B. Interaction of Candida albicans with host cells: Virulence factors, host defense, escape strategies, and the microbiota. J. Microbiol. 2016, 54, 149–169.
- McManus, R.M.; Heneka, M.T. Role of neuroinflammation in neurodegeneration: New insights. Alzheimer’s Res. Ther. 2017, 9, 14.
- Kumar, D.K.; Choi, S.H.; Washicosky, K.J.; Eimer, W.A.; Tucker, S.; Ghofrani, J.; Lefkowitz, A.; McColl, G.; Goldstein, L.E.; Tanzi, R.E.; et al. Amyloid-beta peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci. Transl. Med. 2016, 8, 340ra372.
- Alonso, R.; Pisa, D.; Marina, A.I.; Morato, E.; Rabano, A.; Carrasco, L. Fungal infection in patients with Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 41, 301–311.
- Alonso, R.; Pisa, D.; Fernandez-Fernandez, A.M.; Carrasco, L. Infection of Fungi and Bacteria in Brain Tissue from Elderly Persons and Patients with Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 159.

