1. The Herpesviridae Family
There is considerable evidence implicating herpesviruses in the pathogenesis of Alzheimer’s disease. Herpes simplex virus (HSV) type 1 and type 2 are ubiquitous pathogens that persist for the life of the infected individual. Often, only a certain population of those infected actually show symptoms, while the remainder are asymptomatic [
75]. Herpes simplex virus type 1 (HSV-1) affects the majority of the population, attaining 90% prevalence by the sixth decade of life, with geographic variations. During 2015–2016, in the US alone, the seroprevalence of HSV-1 was 47.8% and the seroprevalence of HSV-2 was 11.9% in the National Health and Nutrition Examination Survey [
76]. While there are eight types of herpesviruses belonging to three subclasses, only seven are implicated in Alzheimer’s disease: HSV-1, HSV-2, VZV, human herpesvirus 6A, human herpesvirus 7, CMV, and EBV.
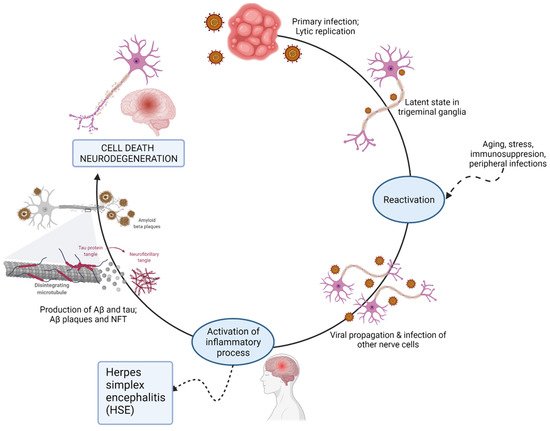
The double-stranded neurotropic virus can establish lifelong latency in nervous tissues, particularly the trigeminal ganglia of the peripheral nervous system, and can reactivate periodically in response to a variety of stimuli, such as immunosuppression, inflammation, peripheral infection, or other stressors [
77]. In 1982, Melvin Ball hypothesized that HSV-1 was causative in AD pathogenesis [
78]. The viral concept of AD proposes that latent HSV-1 located in the trigeminal nerve could reactivate and ascend into areas of the brain. The effect of repeated reactivation, inflammation, and direct viral action may be cumulative and can lead to the development of AD. Herpes simplex encephalitis (HSE) affects the frontal lobes, temporal lobes, and hippocampus [
74]. Interestingly, herpes encephalitis shows cognitive, memory, and behavioral declines analogous to those seen in AD. HSE is frequently associated with epilepsy, which is recognized as a comorbidity in early onset AD patients. Conversely, with APOE4 there is higher epilepsy risk [
79,
80]. Thus, there are functional links between HSE and AD, AD and epilepsy, and consequently, evidence linking HSV-1 to AD.
Recently, studies conducted in a large Taiwanese population yielded results that link antiherpetic medication with a decreased incidence of dementia [
92]. The authors investigated a cohort of roughly 33,000 subjects aged ≥50 years old during the year 2000, who were diagnosed with HSV-1 or HSV-2 infections. The incidence of dementia in the age- and gender-matched control group and the HSV group were investigated for 10 years (2001–2010). The risk of developing dementia in the HSV group was 2.56-fold greater; the main effect was seen in those with HSV-1 infections. Interestingly, a group of HSV-infected patients who had been treated with one of various antiherpetic agents showed a reduction in the incidence of dementia compared to those who received no treatment [
75]. These studies postulate that antiherpetic medications are likely associated with decreased risk of developing dementia and could be used to prevent or slow disease progression. While these studies are promising, there are no data on the effect of antivirals in subjects who are already suffering from AD. Along with the data on HSV-1 presence in elderly brains and its link to APOE-ε4 in AD, these studies support a causal role of HSV-1 in AD [
81,
90].
It is also worth noting that other viruses in the Herpesviridae family, such as HHV-6/7, HHV-2, varicella zoster, and cytomegalovirus, can establish latency and persist for life after the initial infection [
91]. However, studies focused on the Herpesviridae family in the context of age-related diseases are limited. In a recent study, Readhead et al. suggested that HHV-6A and HHV-7 could be causal contributors to AD [
93]. HHV-6A and HHV-7 could induce cell apoptosis in developing thymocytes following primary infection [
94]. The activation of HHV-6A and HHV-7 lytic replication in infected neurons may establish the process by which the viruses cause brain damage [
95]. It has been previously reported that natural killer (NK) cells are activated in response to infection. Thus, indirect effects of the activated host immune system could also destroy brain tissue. Some have theorized that the APP pathway might be hijacked by HHV-6A/7 in the aging brain to cause damage [
95]. The viral induction of AD is depicted in
Figure 4.
Figure 4. Possible mechanisms for virus-induced neurodegeneration.
2. Oral Bacteria
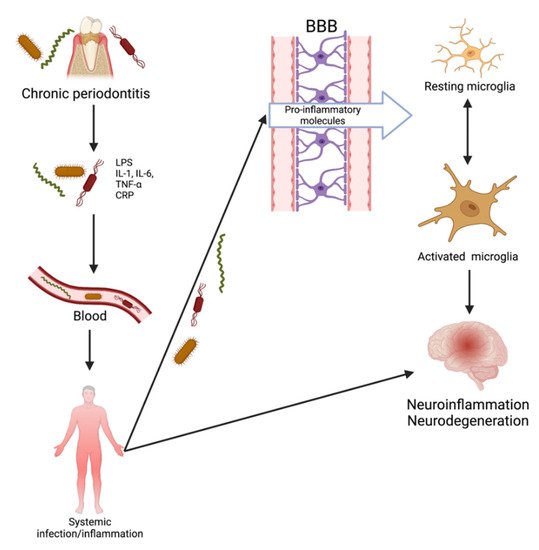
Periodontal disease is an inflammatory dysbiosis with a significant bacterial burden that elicits a systemic inflammatory response and the release of proinflammatory cytokines. There are numerous oral pathogenic bacteria, several of which are invasive, including, but not limited to,
Treponema denticola and
Porphyromonas gingivalis.
Treponema denticola (along with several other oral treponeme species, such as
T. pectinovorum and
T. socranskii) can cause periodontal disease, which, if untreated, can result in edentulism, commonly known as tooth loss. According to the World Health Organization, an estimated 5–20% of adults 65 and older suffer from chronic periodontitis [
96]. Chronic periodontitis has been implicated in various systemic conditions, including rheumatoid arthritis, diabetes mellitus, and more recently, AD. Diseased periodontal pockets host roughly 200 to 300 bacterial species [
97]. The ulcerated lining of the periodontal pockets can reach 20 cm
2, providing bacteria with direct access to the systemic vasculature [
98]. Following entry into circulation, bacterial and inflammatory molecules can access the brain.
There are currently two proposed pathways by which these invasive periodontal spirochetes can reach the brain: inflammatory and bacterial. It has been proposed that chronic periodontitis leads to tissue destruction by inducing an immune response. The interaction between periodontopathic bacteria and host immune cells results in a locally increased production of IL-1β, IL-6, IL-8, and TNF-α. Prolonged exposure of the brain to spirochetal infection and inflammatory mediators “primes” microglia in individuals and results in an inadequate neutralization of invading pathogens reaching the brain. Pro-inflammatory mediators penetrate the BBB, act on the already-primed microglial cells, and trigger the production of Aβ and tau phosphorylation. The resulting neuronal damage could proceed to AD development. Inflammation plays an essential role in periodontitis and, accordingly, has the potential to be causal in AD [
99]. The second mechanism by which the bacteria could contribute to brain inflammation is through bacterial products [
98]. Once in the brain, periodontopathic bacteria that are rich in lipopolysaccharide (LPS) or their products are capable of stimulating cytokine production. Chronic neuronal stimulation by LPS may result in damage to neurons and induce astrocyte activation and glia [
100]. Systemic lipopolysaccharides can quickly increase cytokines within the hippocampus. In the brain, these bacteria and inflammatory molecules can enhance AD-specific pathology, subsequently resulting in neurodegeneration.
Mounting evidence exists for the role of
P. gingivalis in AD, resulting from animal, in vitro, and clinical studies [
118,
119,
120,
121,
122,
123,
124,
125,
126,
127]. Almost all studies have shown neuroinflammation in the brains of experimental animals, but four confirmed the presence of the pathogen DNA and lipopolysaccharides [
119,
120,
123,
127] in the CNS. Similarly, two studies showed neurodegeneration, and four showed cognitive impairment in animal models [
119,
121,
122,
124,
126]. Recently, Ilievski et al. confirmed these results with a periodontal model by showing that
P. gingivalis reached the brain and induced neuroinflammation, AD-specific pathology, and neurodegeneration [
120]. Dominy et al. found that when added peripherally, inhibitors of gingipains (trypsin-like cysteine proteinases that can degrade cytokines) were able to counteract the pathological effects of
P. gingivalis in the brain [
119]. These studies show that the periodontal bacterial species are able to induce brain pathology, cognitive impairment, neurodegeneration, blood–brain barrier disruption, and neuroinflammation (
Figure 5). Altogether, these various studies provide support for the theory that periodontitis can occur before AD pathology and that it has the potential to induce Alzheimer’s disease pathophysiology.
Figure 5. Transition of periodontitis to neurological disease.
3. Borrelia burgdorferi
Spirochetes are corkscrew-shaped bacteria that are the causative agents for several chronic diseases including syphilis, Lyme disease, and periodontal disease. Because spirochetes are strongly neurotropic, there is an increasing amount of data that indicate that spirochetal infection causes Aβ deposition and plaque- and tangle-like lesions and, therefore, might be involved in dementia and AD pathogenesis. Spirochetes disseminate as individual bacteria to the cerebral cortex and may form plaques. Spirochetes evade host defenses and sustain chronic infection and inflammation, which may explain the slowly progressive course of dementia in AD [
128]. The spirochetal aggregates appear similar in morphology to cortical senile plaques in AD [
129]. Histochemical, immunohistochemical, and in situ hybridization techniques further demonstrated that Aβ and bacterial DNA are vital components of both pure spirochetal aggregates and senile plaques [
128]. Researchers have reported that the number of spirochetes and plaques in the hippocampus and frontal cortex increases with the severity of cortical atrophy [
130]. Previously, researchers have suggested that APP is an integral part of spirochetes, indicating that bacterial amyloid could contribute to senile plaque formation [
67].
One spirochete that has been implicated in the etiology of Alzheimer’s disease is
Borrelia burgdorferi.
B. burgdorferi, a bacterium transmitted to humans through the bite of an
Ixodes tick, is the etiologic agent of Lyme disease (LD). Lyme disease is characterized by erythema migrans, extreme fatigue, myocarditis, oligoarthritis of joints, and neurologic dysfunction. The most dangerous outcome of
B. burgdorferi infection is associated with an invasion of the nervous system by the spirochete. Each year in the United States >30,000 cases are reported to the Centers for Disease Control and Prevention (CDC), but a recent estimate using other methods suggests the number of infections is closer to 476,000/year [
131,
132]. The organ pleiotropism of
B. burgdorferi results in diverse manifestations, including neuroborreliosis [
133], which occurs in approximately 10–15% of patients. CNS infection resulting in neurologic Lyme disease is well-documented [
134]. The treatment of LD with oral or intravenous (for late stage) antibiotic therapy, in a majority of cases, leads to an improvement in symptoms after several weeks or months. However, a proportion of patients continue to experience symptoms, a phenomenon known as post-treatment Lyme disease (PTLD), which is considered by some to be a psychosomatic neurocognitive impairment [
135]. However, animal studies have clearly demonstrated the persistence of the LD spirochete in multiple organs, including the brain, after antibiotic treatment [
136,
137,
138]. Prior studies of PTLD patients showed immune activation in both CSF and serum. The activation of the inflammatory response in Lyme neuroborreliosis contributes to the pathogenesis of a broad spectrum of neurologic disorders. The clinical manifestations of neuroborreliosis may include ataxia, Parkinson-like symptoms, cognitive impairment, mental health disorders, paraparesis, and confusion. The discordant findings regarding the successful detection of the spirochetes involved in affected tissues has resulted in a stark difference of opinions. However, given the neurotropic effects of LD, many researchers have speculated a causal association between Lyme disease and neurodegenerative disorders.
While many researchers remain skeptical, there is strong evidence that shows
B. burgdorferi can evade the host immune reactions and cause chronic infection, which further develops into neurological damage. The resultant inflammatory state leads to abnormal tau phosphorylation, microtubular dysfunction, and neurofibrillary tangle generation [
139]. The pathogenic bacteria
Borrelia have been shown to increase the permeability of the blood–brain barrier for entry, causing pleocytosis in the CSF, with white blood cell migration increasing as well [
139]. Spirochetes then disseminate as individual bacteria to the cerebral cortex, and they are thought to form masses or plaques.
B. burgdorferi spirochetes have been shown to produce amyloid deposits and tau hyperphosphorylation, indicating that bacteria and/or their degradation products may enhance the cascade of events leading to dementia.
In 1987, MacDonald and Miranda first reported the incidence of
B. burgdorferi in the brains of AD cases [
68]. The identification was validated using serological methods and morphological and immunohistochemical features. Several other researchers have reported similar findings. Fallon and Nields revealed the association of dementia and microgliosis with cortical atrophy in LD [
140]. Meer-Scherrer et al. detected an association between
B. burgdorferi DNA and neuropathology in the post-mortem brain tissue of a PTLD patient [
140], and Miklossy detected spirochetes in the blood, CSF, and brain tissue of AD cases [
141]. In addition, Miklossy and colleagues discovered that in AD patients NFTs were co-localized with Aβ plaques, and both contained
B. burgdorferi-specific DNA [
128]. Miklossy then examined 147 AD patients for the isolation of spirochetal species by culturing their cerebral cortex and blood in BSK II (Borrelia growth medium) [
142]. In this study, the authors claim to have confirmed the presence of spirochetes in the blood, cerebral cortex, and CSF of 14 AD patients [
129]. The bacterial isolates were further investigated by in situ hybridization and histopathology.
Nonetheless, several studies found no evidence to suggest that
B. burgdorferi is linked to neurodegenerative disorders. Pappolla et al. tested post-mortem brain tissue samples from both AD cases and controls by culturing for Borrelia, but all were negative for growth [
144]. In a similar approach, Gutaker et al. found no evidence of spirochetal infection in 10 postmortem AD brain samples, tested with standard and nested PCR, ELISA, and Western blotting for anti-
B. burgdorferi antibodies [
145]. Marques et al. also used PCR but did not detect
Borrelia in any of the 30 postmortem AD and sex-matched control brain tissue samples [
146]. Interestingly, McLaughlin et al. tested for spirochetes in the peripheral blood and fresh postmortem brain specimens of 22 patients with AD and 6 controls, and only 1 tested positive [
147]. A large case-control study by Galbussera et al. found no evidence of
B. burgdorferi in any serum samples using ELISA from 50 patients with AD, 23 controls, or 25 healthy caregivers of patients with AD [
148]. In a nationwide population-based cohort study in Denmark, researchers observed no increased long-term risk of AD coincident with LD. This study is consistent with a geoepidemiology study from the US in which there were no associations between the geographical distribution of Lyme disease and the geographical distribution of AD [
149]. It should be noted that Lyme disease is frequently associated with other tick- and non-tick-transmitted co-infections, suggesting that concurrent infections with several pathogens may also occur in Alzheimer’s disease. The most important of these co-infections are caused by
Bartonella species,
Babesia species,
Mycoplasma pneumoniae, Yersinia enterocolitica, C. pneumoniae, HSV-1, and
C. trachomatis. Co-infections with these pathogens may exacerbate Lyme disease symptoms through immune system modulation. Given the mixed findings, the etiologic role of
Borrelia burgdorferi in the pathogenesis of AD remains unresolved.
4. Chlamydia pneumoniae
Chlamydia pneumoniae is an obligate, intracellular respiratory pathogen that can persist as a chronic infection for long periods of time.
C. pneumoniae is responsible for a significant proportion of community-acquired pneumonia and has been linked to numerous other pulmonary diseases. Interestingly,
C. pneumoniae has been associated with a range of non-respiratory diseases, including atherosclerosis, inflammatory arthritis, multiple sclerosis, and others [
150,
151,
152]. Several authors reported an association between
C. pneumoniae and AD since the bacteria has tropism for neural tissue. According to Shima and colleagues,
C. pneumoniae could be a trigger for late-onset AD and is the most plausible of all infectious bacterial agents linked to AD pathogenesis [
153]. In particular, the entry of the organism into the human brain is thought to occur following exposure in the respiratory tract. One potential route follows an intracellular infection of the neuroepithelia in the nasal airway to the olfactory bulb and then deeper into brain structures [
154]. The other route would be following uptake in the lung by monocytes, which then traffic the organisms through the vasculature until they reach the brain through the BBB [
154].
Immunohistochemical analyses of AD brains showed
C. pneumoniae in various cell types found in the brain, including endothelial cells, astrocytes, microglia, and neurons [
155,
156,
157,
158,
159,
160,
161]. The pathogen may reside in an intracellular inclusion/vacuole that resists immune recognition. These bacteria require cholesterol and sphingomyelin, which are gathered from the host; therefore,
C. pneumoniae has the ability to manipulate the host cells [
158]. It is proposed that
C. pneumoniae and related antigens may interact with soluble oligomeric forms of amyloid in the same cortical regions of the brains of AD patients [
1]. Currently, anti-chlamydia antibodies on the frontal and temporal cortical sections of AD brains are used to provide insight into the relationship between pathology and infection [
158].
By employing a variety of techniques, including highly specific RT-PCR, immunoelectron microscopy, PCR, and in vivo cell culturing, researchers were able to localize infection in the AD brain. The areas found to be infected were the amygdala, entorhinal cortex, hippocampus proper, and the temporal and frontal cortices. Immunoelectron and electron microscopy identified chlamydial elementary bodies and reticulate bodies (RBs) [
155,
161]. The replicative RB form was detected in neurons, pericytes, and glial cells, which indicates that a viable and active form of
C. pneumoniae is present in these cells [
156,
162,
163,
164]. In addition, Balin et al. found that 90% of AD brains, particularly the cerebral region, were found to be PCR-positive for the pathogen [
155]. Schumacher et al. identified DNA of the organism in 90% of postmortem brain samples from LOAD patients and 5% of non-dementia control samples [
20,
150].
C. pneumoniae DNA was detected in the CSF of 43.9% of AD patients compared to 10.6% of the controls [
165]. These findings suggest that
Chlamydia pneumoniae infection in the brain should be considered a triggering agent in the initiation of AD pathogenesis. Other studies failed to detect
C. pneumoniae DNA in the tissue of AD patients [
20]. It should be noted that two of these studies were performed on paraffin-embedded tissues, which may have affected researchers’ ability to amplify DNA using the PCR technique [
166,
167].
2.5. Fungal Pathogens
Recently, researchers have advanced the idea that disseminated yeast and fungal infections contribute to the progression of AD. Under normal circumstances,
Candida spp. live in the human intestinal tract along with other species of bacteria and yeast as a natural part of our microbial flora.
Candida albicans is the most prolific cause of fungal infections in humans. The CDC estimates that roughly 25,000 cases of candidemia occur each year in the United States [
168].
C. albicans is a commensal fungus that can readily infect organs and colonize inside resident cells and macrophages as endomycosomes or phagosomes [
169]. The fungal AD hypothesis refers to the Carrasco et al. fungal etiology based on AD autopsies that associated fungi and neural tissue [
170,
171,
172,
173,
174]. In the fungal model, a defeat of the innate immune system allows for the colonization of neural cells with fungi. Like a trojan horse,
Candida spp. invade endothelial cells by endocytosis and escape the immune system by residing in endomycosomes. Fungi may enter into the cranial cavity via the nasopharyngeal nerve complex and then into the olfactory bulb, a region implicated in the development of dementia [
175]. The fungi prevent removal by the immune system by maintaining low levels of colonization, which avoids activating apoptotic pathways [
176]. As fungal burden increases, hyphal development induces cytokine production, which recruits neutrophils and macrophages. As the immune response clears the fungal burden, the colony falls from a virulent state to a low level of colonization [
177]. Thus, researchers hypothesize that disseminated infection may slowly spread to the CNS, and neuronal loss takes place only when the fungal burden is high [
170].
It has been suggested that Aβ functions as an antimicrobial peptide. Interestingly,
C. albicans was found to be sensitive to synthetic Aβ and brain homogenates from AD patients that were capable of inhibiting fungal growth [
178]. Additionally, it was demonstrated that Aβ protects against
C. albicans in glial cells as well as in vivo in nematodes [
179]. Therefore, the formation of Aβ is a response by the innate immune system in an attempt to protect against infection in the brain. Based on the assumption that fungi are the etiological agent of AD, all symptoms observed in the disease can readily be explained. The slow progression of Alzheimer’s disease reflects the chronic nature of fungal infections, if left untreated.
Alonso and colleagues provided extensive evidence that disseminated mycoses are potential causative agents or risk factors for AD [
173,
180]. Different fungal genera detected in AD brain tissue include
Malassezia, Fusarium, Candida, Cladosporium, Alternaria, and
Botrytis [
181]. An analysis of CSF revealed significant levels of
Candida albicans and
Candida glabrata in samples from AD patients. Approximately 89.6% of serum from AD patients tested positive for antibodies to
Candida compared to 8.8% for controls [
173]. These studies revealed that many AD patients could have been co-infected with a variety of fungal species, while no fungal DNA was found in control samples. The different species that were detected included
Saccharomyces cerevisiae,
Malassezia globosa,
Malassezia restricta, and
Penicillium. Furthermore, this group detected yeast and fungal proteins, including (1,3)-β-glucan, fungal polysaccharides, and mycoses, in the peripheral blood of AD patients, which suggests that a chronic fungal infection may increase the risk of dementia [
173,
180]. More strikingly, yeast-like cells and hyphal structures were observed in CNS tissue from AD patients using polyclonal antibodies against a variety of fungi [
181]. Pisa et al. also provided strong evidence for fungal infection in AD patients [
170]. Brain sections derived from AD patients showed that all were infected with fungi [
170]. Fungal material was detected intra- and extracellularly in the neurons of tissues of AD patients, but no fungal material was observed in tissue from control individuals [
170]. Moreover, fungal DNA and proteins were also found in brain regions including the frontal cortex, hippocampus, cerebellar hemisphere, and choroid plexus but not in control patient tissue. Immunohistochemistry and confocal microscopy using anti-
C. glabrata antibodies detected fungal bodies inside and outside nuclei and, in some cases, contained fungal nucleic acid [
170]. The possibility that AD is a fungal disease opens new perspectives for treatment and therapy for patients. The aforementioned findings demonstrate that fungi can be detected in the CNS of AD patients and support the possibility that fungal infections are linked to AD.
This entry is adapted from the peer-reviewed paper 10.3390/neurosci3020019