Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Diego Estrada Luna | -- | 3535 | 2022-05-31 16:25:42 | | | |
| 2 | Peter Tang | Meta information modification | 3535 | 2022-06-01 02:57:07 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Estrada Luna, D.; Jiménez-Osorio, A.S.; Jaen Vega, S.; Fernández-Martínez, E.; Ortiz Rodriguez, M.A.; , .; Flores-Chávez, O.R.; Ramírez-Moreno, E.; Arias-Rico, J.; Arteaga-García, F.; et al. Antioxidant Phytochemicals in HIV+ Patients. Encyclopedia. Available online: https://encyclopedia.pub/entry/23610 (accessed on 09 August 2026).
Estrada Luna D, Jiménez-Osorio AS, Jaen Vega S, Fernández-Martínez E, Ortiz Rodriguez MA, , et al. Antioxidant Phytochemicals in HIV+ Patients. Encyclopedia. Available at: https://encyclopedia.pub/entry/23610. Accessed August 09, 2026.
Estrada Luna, Diego, Angélica Saraí Jiménez-Osorio, Sinai Jaen Vega, Eduardo Fernández-Martínez, Maria Araceli Ortiz Rodriguez, , Olga Rocío Flores-Chávez, Esther Ramírez-Moreno, José Arias-Rico, Felipe Arteaga-García, et al. "Antioxidant Phytochemicals in HIV+ Patients" Encyclopedia, https://encyclopedia.pub/entry/23610 (accessed August 09, 2026).
Estrada Luna, D., Jiménez-Osorio, A.S., Jaen Vega, S., Fernández-Martínez, E., Ortiz Rodriguez, M.A., , ., Flores-Chávez, O.R., Ramírez-Moreno, E., Arias-Rico, J., Arteaga-García, F., & Estrada-Luna, D. (2022, May 31). Antioxidant Phytochemicals in HIV+ Patients. In Encyclopedia. https://encyclopedia.pub/entry/23610
Estrada Luna, Diego, et al. "Antioxidant Phytochemicals in HIV+ Patients." Encyclopedia. Web. 31 May, 2022.
Copy Citation
Human immunodeficiency virus (HIV) infection has continued to be the subject of study since its discovery nearly 40 years ago. Significant advances in research and intake of antiretroviral therapy (ART) have slowed the progression and appearance of the disease symptoms and the incidence of concomitant diseases, which are the leading cause of death in HIV+ persons.
antiretroviral therapy
phytochemicals
lipid metabolism
genes
HIV
oxidative stress
1. Introduction
Since the first case was reported in the 1980s, human immunodeficiency virus (HIV) infection has spread worldwide and become one of the leading causes of death [1]. In 2020, it was estimated that approximately 37.6 million people were infected with HIV. A total of 95.5% of them were adults, and 4.5% were children under 14 years of age; on account of the low accessibility of antiretroviral therapy (ART), children comprise the most vulnerable group to develop a comorbidity or die. Treatment with ART is critical at the initial stages of infection because it reduces the virus replication rate and keeps the viral load at undetectable levels [2]. The main antiretroviral drugs currently available and the most effective have been described as protease inhibitors (PIs), integrase inhibitors (INIs), and reverse transcriptase inhibitors (RTIs), which are divided into nucleoside analogs (NARTI or NRTI), nucleotide analogs (NtARTI or NtRTI) and non-nucleoside analogs (NNRTI) [3]. However, the use of these antiretrovirals may increase oxidative stress (OS) because, together with the natural course of the disease, an increase in reactive oxygen species (ROS) and other prooxidant substances have been found in the organism [4][5][6]. OS may activate pro-inflammatory signaling pathways, considered risk factors for various chronic degenerative diseases [7][8], the main ones linked to cardiovascular events [9][10][11]. Likewise, OS significantly decreases the effectiveness of ART, enhancing the complication of the illness and the appearance of opportunistic conditions [12].

2. HIV
HIV is a retrovirus of the lentivirus genus that causes a slow and progressive reduction of the immune system due to viral replication, mainly in the CD4 lymphocyte cells [13].
After entering the body, the virus infects host cells by binding to the CD4 receptor, and chemokine CCR5 or CXCR4 co-receptors found mainly on T lymphocytes (Figure 1) and macrophages, dendritic cells, and monocytes. Inside the cell, single-stranded RNA is released, which will serve as a template for synthesizing double-stranded viral DNA by retrotranscription, allowing the virus to enter the nucleus helped by integrase to place its genetic material with that of the host cell. Once the new viral RNA is formed, it is used as genomic RNA to form viral proteins that will be mobilized back to the cell membrane, giving rise to an immature (non-infectious) virus; this leads to the release of proteases from the virus for the degradation of the long-chain polypeptides, generating mature viruses that will allow the virus to spread in the organism [14][15][16].
Figure 1. Lifecycle of HIV: binding, fusion, reverse transcription, integration, replication, assembly, budding, and maturation.
HIV infection is branded by a series of stages with peculiar clinical evidence, such as the acute retroviral infection phase, which may be described as asymptomatic, although it can be accompanied by pharyngitis, fever, myalgias, and others. Likewise, gastrointestinal, dermatological, and neurological symptoms may also occur [17][18]. The asymptomatic phase is marked by being asymptomatic or presenting an adenoid syndrome with the presence of firm, but not woody, mobile, non-painful lymph nodes without changes in the overlying skin, occupying two or more adjacent regions [19][20]. During the AIDS phase, immunosuppression is exacerbated with a considerable loss of CD4 lymphocytes and significant viral replication, neoplasms, and opportunistic infection [21][22][23][24][25][26][27].
The initial treatment of HIV is essential to avoid the progression of the infection. However, only 59% of people with HIV receive ART and follow up the illness in a care center; other people abandon the service after being diagnosed and return when the illness is significantly advanced and has deteriorated the immune system; which translates into a significant increase in morbidity and mortality rates [28][29].
2.1. Antiretroviral Therapy (ART)
ART has increased the life expectancy in HIV patients such that it is similar to that of the general population, making HIV a treatable chronic disease from a multidisciplinary approach [22][30].
One of the most effective combinations is highly active antiretroviral therapy (HAART), where three or more drugs are used to suppress the viral load to undetectable levels, leading to immunological recovery in a shorter time. It is considered the most effective strategy for treating HIV and also concerning its cost–benefit ratio, since it reduces hospitalizations due to complications, and the incidence of opportunistic infections, improving the quality of life [31][32].
2.2. Effects of Antiretroviral Therapy on Lipid and Cholesterol Metabolism
In addition to HIV infection, the administration of PI-based ART or combined ART and the inflammation that occurs in the disease leads to changes in lipid metabolism that are associated with a rise in the incidence of metabolic risk factors, including insulin resistance, dyslipidemia, glucose intolerance, metabolic syndrome (MetS), lactic acid elevation, and some types of lipodystrophy [33][34][35][36][37][38]. As the disease progresses, CD4 lymphocytes induce the inflammatory response by increasing the levels of OS and the production of cytokines, which results in a decrease in the concentration of high-density lipoprotein cholesterol (HDL-C), altering the reverse cholesterol transport (RCT) and accumulating cholesterol in macrophages [39]. On the other hand, the proliferation of pro-inflammatory lipids as oxidized low-density lipoproteins (ox-LDL) are associated with the expression of inflammatory markers of the immune system as interleukin 1β, tumor necrosis factor alpha (TNF-α), and interleukin six, and that lasts even if viral replication is inhibited by ART. This pro-inflammatory state correlates with the development of thrombosis. Besides this, the increase in viral proteins and cytokines stimulates endothelial lipase activity leading to mitochondrial dysfunction, the production of prooxidant chemical species, insulin resistance, a decrease in adiponectin, and an increment in remnant lipoproteins [40].
In the same way, it has been established that HDL-C, both large (HDL2b and HDL2a) and small particles (HDL3a, HDL3b, and HDL3c), have been associated with cardiovascular risk in HIV patients. Although this process is exacerbated by ART, it continues to be unclear whether regulation of its chemical composition can modify changes in lipid metabolism and the effects produced by ART, which include rising OS and activation of pro-inflammatory pathways [41][42]; however, the increase in plasma HDL-C concentration does not reverse cardiovascular risk.
2.3. Effects of HIV Infection and ART on Lipid and Cholesterol Genes
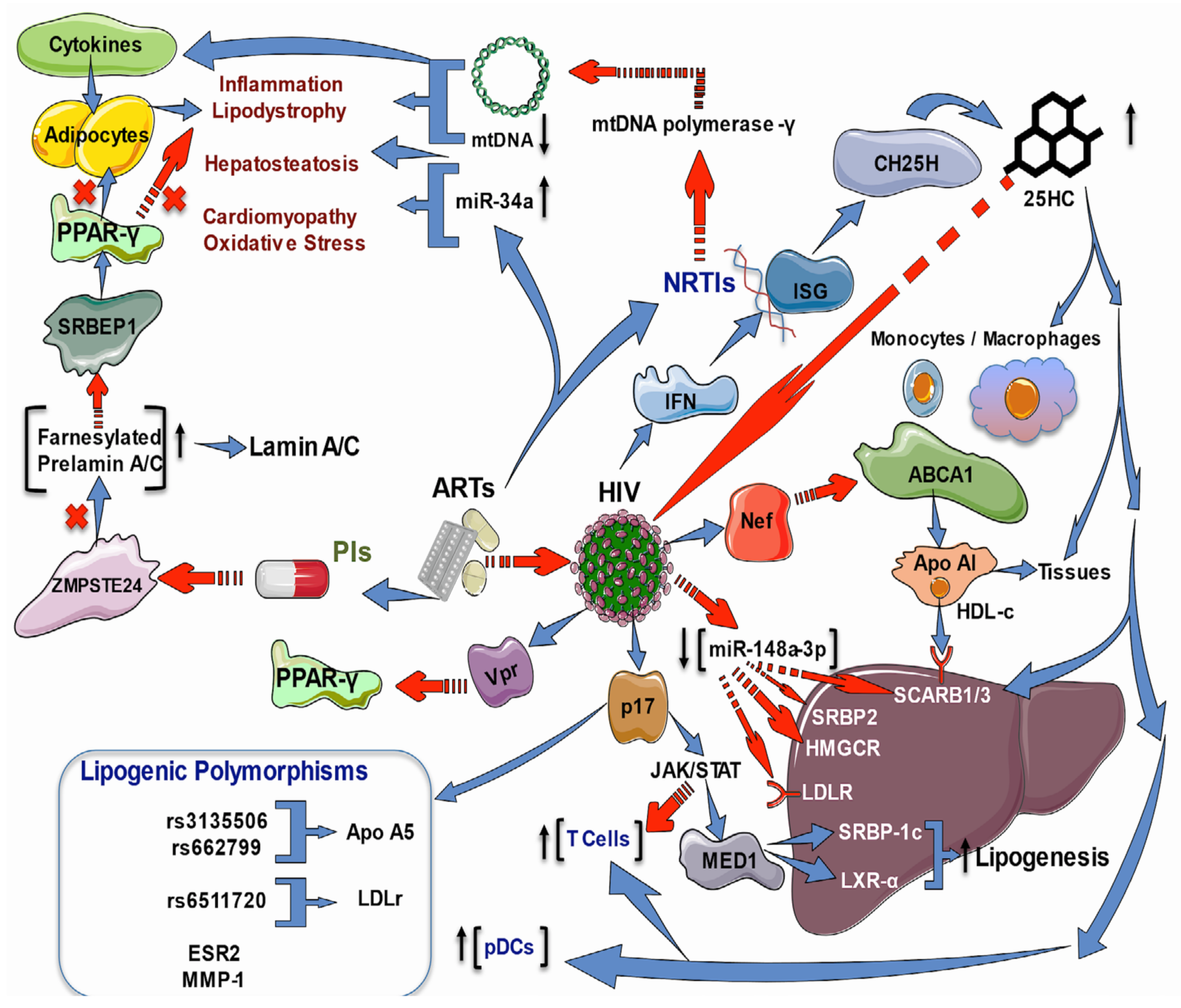
Lipodystrophies are characterized by differing degrees of body fat loss, including a tendency to metabolic disturbances such as insulin resistance, diabetes, hypertriglyceridemia, and hepatic steatosis; their origin can be genetic or acquired. The two most common acquired types are generalized and partial lipodystrophy. Common subtypes are HAART-associated lipodystrophy syndrome (HAALS) in HIV patients and drug-induced localized lipodystrophy. Indeed, HAALS seems to be multifactorial and may occur after two or four years of HAART in patients administered with PIs or NRTIs [43]. As PIs inhibit the zinc metalloprotease STE24 (ZMPSTE24), the enzyme responsible for prelamin A processing, an accumulation of toxic farnesylated prelamin A may cause the dysregulation of transcription factors involved in adipogenesis and HIV-associated cardiomyopathy provoked by inflammation—perhaps due to the modulation of NFκB signaling via the DNA damage transducer ataxia telangiectasia mutated (ATM) [43][44]. Moreover, NRTIs inhibit the mitochondrial DNA (mtDNA) polymerase-γ transcription, leading to mtDNA depletion, which causes diverse pathologies, including lipodystrophy and hepatosteatosis mediated by pro-inflammatory cytokines [45]. These data suggest that targeting inflammation may be a helpful treatment approach.
Not only do the drugs used in ART cause metabolic disturbances, but HIV infection alone has also been strongly associated with an increased risk of cardiovascular diseases. Untreated HIV-infected persons present lower levels of HDL-C than HIV-negative people; HDL-C levels are lower after the acquisition of the virus [46][47]. It has been demonstrated in apoE−/− mice that the accumulation of abnormal metabolites such as oxLDL upregulates purinergic 2X7 receptor (P2X7R), nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3) inflammasome, and interleukin (IL)-1β expression during atherosclerotic plaque formation through protein kinase R (PKR) phosphorylation [48][49]. In HIV infection, HIV-1 transactivator of transcription protein (Tat) and HIV-1 protease increase NLRP3 inflammasome [50] and contribute to CD4+ T loss through pyroptosis [51]. Additionally, hypercholesterolemia increases caspase-1 activity in the coronary arterial endothelium of Nlrp3(+/+) mice through superoxide production, leading to the downregulation of endothelial nitric oxide synthase activity pyroptosis [52].
HDL-C also participates in the RCT from peripheral tissues into circulation and the liver, where cholesterol can be metabolized or eliminated [53]. It has been demonstrated in vitro that HIV infection via negative regulatory factor (Nef) protein harms the monocyte-macrophage cholesterol efflux by increasing the degradation of the ATP-binding cassette transporter-A1 (ABCA1), a crucial transporter of lipids and cholesterol from cells to extracellular apolipoprotein-A1 [53], but by up-regulating its mRNA [54]. In vivo, untreated HIV persons had lower HDL-C levels while ABCA1 mRNA was elevated compared to ART-treated patients and HIV-negative people.
Interestingly, the expression of genes related to cholesterol uptake (low-density lipoprotein receptor, LDLR, and scavenger receptor class B member-3, SCARB3, also known as cluster of differentiation-36, CD36), synthesis (3-hydroxy-3-methylglutaryl-CoA reductase, HMGCR), and regulation (sterol regulatory element-binding transcription factor 2 or SREBP2 and liver X receptor-alpha, LXRα) were significantly decreased in treated or untreated HIV infected patients in contrast to the HIV-negative group [47]. That study indicates that HIV and ART impact monocyte-macrophage cholesterol metabolism, which is vital for forming foam cells and atherosclerosis. Furthermore, a study on HIV treatment-experienced individuals revealed a dysregulation between SRBP2 and HMGCR and LDLR pathways, which may precede the clinical manifestation of ART-induced lipid metabolism derangement [55].
On the other hand, interferons (IFN) are recognized antiviral cytokines which up-regulate many interferon-stimulated genes (ISG). One of these ISG is cholesterol-25-hydroxylase (CH25H), which converts cholesterol to 25-hydroxycholesterol (25HC), a soluble antiviral factor since, in cultured cells, 25HC inhibits the growth of enveloped HIV by blocking membrane fusion with cells; besides, in humanized mice, the 25HC suppressed the HIV replication and T cell depletion [56]. Furthermore, there is strong evidence that sterol metabolism may confer natural resistance to HIV-1 infection in exposed seronegative people (HESN). For instance, peripheral blood mononuclear cells (PBMCs) and monocyte-derived macrophages (MDMs) were isolated from HESN and compared to healthy controls; also, MDMs from five healthy controls were in vitro HIV-1-infected in the absence or presence of 25HC. IFN-producing plasmacytoid dendritic cells (pDCs) were augmented in HESN than healthy controls in unstimulated and in vitro HIV-1-infected PBMCs. The expression of CH25H and several genes involved in cholesterol metabolism (ABCA1, ABCG1, cytochrome P450 family 7 subfamily b member 1, CYP7B1, LXRα, oxysterol binding protein, OSB.P., peroxisome proliferator-activated receptor-gamma, PPARγ, and SCARB1) were increased. These results were correlated with reduced susceptibility to in vitro HIV-1-infection of PBMCs and MDMs. Remarkably, the 25HC added to MDMs caused an increased cholesterol efflux and augmented resistance to in vitro HIV-1-infection [57].
Metabolic comorbidities of both HIV infection alone and ART-induced ones include inflammation, dyslipidemia, atherosclerosis, MetS, lipodystrophy, myocardial disorders, diabetes, impaired hematopoiesis, cognitive damage, and liver injury. As previously commented, most of these depend on the cholesterol metabolism, which is altered by the HIV infection process itself and can be enhanced by HAART. Currently, there is a growing body of evidence pointing out the crucial role of Nef-induced impairment of genes involved in the cholesterol metabolism and the cholesterol-enriched regions of the plasma membrane, known as lipid rafts, which facilitate the HIV replication and the successful entry/exit of HIV in the target cells [58]. Nevertheless, Nef is not the only protein involved in the metabolic disorders observed in HIV patients; the HIV protein Vpr inhibits the PPARγ leading to lipotoxicity in mouse models [59] or, as mentioned above, the protein p17 that mediates liver steatosis. Another comorbidity is alcohol use in HIV patients. Because the in vitro and in vivo oxidative stress provoked by alcohol reduced the expression of antioxidant enzymes such as glutathione synthetase (GSS), superoxide dismutase (SOD), and glutathione peroxidase (GPx), as well as lowered ABC-transporters and the SREBP-2 transcription; while increased membrane lipid rafts caveolin-1 (Cav-1), and up-regulated HMGCR and cyclooxygenase-2 and 5-lipoxygenase (5-LOX), resulting in high levels of pro-inflammatory prostaglandin-E2. In sum, oxidative stress exacerbates the injury caused by HIV infection in alcohol consumers (Figure 2) [60].
Figure 2. Effects of ART and HIV infection on lipid and cholesterol regulatory genes. ART may cause lipodystrophy in HIV patients administered with PIs or NRTIs. PIs inhibit ZMPSTE24, which processes the farnesylated prelamin-A/C, inducing its accumulation. Prelamin-A/C sequesters S.R.E.B.P., decreasing its activity on PPAR-γ, impairing the regulation of adipogenesis transcription, and promoting the HIV-associated cardiomyopathy by NF-kB-induced inflammation. NRTIs inhibit mtDNA polymerase-γ transcription, leading to mtDNA depletion, which causes lipodystrophy and hepatosteatosis mediated by pro-inflammatory cytokines. Besides this, ART increases miR34a, promoting hepatosteatosis, cardiomyopathy, and OS HDL-C with Apo1 participates in the RCT from peripheral tissues into circulation and the liver. HIV infection via Nef harms the monocyte-macrophage cholesterol efflux by increasing ABCA1 degradation; also, the downregulation of genes related to cholesterol uptake (LDLR and SCARB1/3), synthesis (HMGCR), and regulation (SREBP2 and LXRα). The HIV matrix protein p17 enhances the expression and transcriptional activity of LXR, and its coactivator (MED1), via the activation of Jak/STAT signaling, which results in hepatic lipid accumulation via activation of the LXR/SREBP1c lipogenic pathway and mediates liver steatosis. HIV-positive patients on ART present gallstones and higher total cholesterol with significantly elevated LDL-C levels but decreased scavenging LDLR for LDL-C. The transcriptional regulator of LDLR, SREBP2, is decreased in HIV infection; besides this, the regulatory miR-148a-3p is reduced with a concomitant increase in target ABCA1. Additionally, the HIV protein Vpr inhibits the PPARγ leading to lipotoxicity. Antiviral IFNs upregulate ISGs (CH25H), which converts cholesterol to 25HC that inhibits the growth of enveloped HIV by blocking membrane fusion with cells, suppresses the HIV replication, and increases the number of T cell and pDCs; it also augments the expression of genes involved in cholesterol metabolism (ABCA1, ABCG1, CYP7B1, LXRa, OSB.P., PPAR-γ, and SCARB1/3). The SNPs rs3135506 and rs662799 of the APOA5 gene and rs6511720 of the LDLR gene were associated with the development of atherogenic dyslipidemia. The T allele of ESR2 and G.G. genotype of MMP1 were found to be associated with lipoatrophy.
2.4. Increased Oxidative Stress in People HIV+
OS is understood as the imbalance between the production of ROS/reactive nitrogen species (RNS) and other prooxidant chemical species and the antioxidant system in the body. ROS and RNS affect the cytochrome p450 enzyme system [61], required for chemical defense or detoxification, and the cellular response system of molecular signals [62]. Awareness of chemical prooxidant species and antioxidants has been gaining interest in recent decades because of their significant role in different signaling pathways, cell homeostasis maintenance, phagocytosis processes, immunological functions, vascular processes, and cell membrane stability [63][64]. ROS are generated naturally by aerobic cellular metabolism when oxygen is partially reduced, producing hydrogen peroxide (H2O2), superoxide anion (O2•–), free oxygens (1/2O2), and hydroxyl radicals (OH•). Species from nitrogen, iron, copper, and sulfur substances are equally found [65][66]. When the levels of prooxidant species are not removed or processed by the endogenous antioxidant system, oxidative processes are activated and damage biomolecules such as proteins, carbohydrates, lipids, and DNA, being factors for the development of cardiovascular diseases, diverse types of cancer, neurological conditions, and other diseases featured by the presence of inflammatory processes [67][68].
Other frequent forms of ROS production are through the electron transport chain, degradation of lipids and amino acids, and protein folding in the lumen of the endoplasmic reticulum (ER). ROS production is mediated by the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) family complex as NOX1, NOX2, NOX4, and NOX5 [5][69]. NOX members are found in muscle tissue and represent the primary sources of ROS; its activity has been investigated in conditions such as hypertension, hypercholesterolemia, and endothelial dysfunction [70][71][72][73][74][75]. Another enzyme that generates ROS is xanthine oxidoreductase, principally found in the liver and gut, although distributed in smaller proportions in the lungs, kidneys, heart, and brain plasma. Among its functions are redox reactions of sulfhydryl groups through intrasubunit disulfide bridges that allow the conversion of NAD+-dependent xanthine dehydrogenase (XDH) into oxygen-dependent xanthine oxidase (XO). Furthermore, both XO and XDH can oxidize NADH with the concomitant formation of ROS [5][69].
Given these conditions, natural therapies based on foods with high antioxidant capacity seem to represent an efficient option to prevent and diminish the organic functional deterioration caused by the OS excess generated by HIV and ART.
2.5. Antioxidants and Phytochemicals
Antioxidants are substances present at low concentrations compared to those of an oxidizable substrate, which remove, retard, or prevent the oxidation of the substrate. Antioxidants, when interacting with a free radical, yield an electron to it, oxidizing it in turn and transforming it into a relatively stable, non-toxic molecule which, in some cases, can be regenerated to its reduced form by the action of other antioxidant systems (for example vitamin E) [76][77].
The nature and structure of antioxidants are diverse, and they are traditionally classified into endogenous and exogenous antioxidants. There are those belonging to the superoxide dismutase (SOD) family among the endogenous antioxidants, such as manganese SOD (MnSOD), the only enzyme of this family found inside the mitochondria [78], copper-zinc SOD (CuZnSOD), which acts in cytoplasm reducing superoxide anion [79], and extracellular SOD (ecSOD), the principal regulator of nitric oxide (NO) in the vasculature [80]. Other essential antioxidant enzymes are catalase (CAT), which promotes the breakdown of H2O2 and sulfur oxidase or sulfur reductase activity [81], selenium-dependent glutathione peroxidase (SeGPx); also, the thioredoxin system, an NADPH-dependent enzyme, thioredoxin reductase (TrxR) implicated in DNA and protein repair [82]. In addition, TrxR serves as a scavenger of transition metals (iron, copper, and silver) and non-enzymatic metabolites with antioxidant capacity (GSH, urate, bilirubin, ubiquinones). Exogenous antioxidants enter through the food chain and require continuous renewal. Natural exogenous antioxidants include vitamins (E and C), lipoic acid, selenium, phytochemicals β-carotenes, vitamin A, ellagic acid, and flavonoids [83].
Among synthetic antioxidants are the transition metal chelators (deferoxamine, α-keto-hydroxy-pyridines), ROS scavengers as 21-amino-steroids, 2-methyl-aminochromanes, pyrrolopyrimidines, butylated hydroxytoluene, phenyl-tert-butyl-nitrone, n-acetyl-cysteine, nonsteroidal anti-inflammatory drug, probucol, β-blockers, calcium-channel blockers, angiotensin-converting enzyme inhibitors, and xanthine oxidase inhibitors (allopurinol), antioxidant enzymes for therapeutic use (pyran-SOD, desferal/MnIII, Fe-TPEN, Fe-TPAA, EUK-8, M40403, ebselen), NADPH oxidase inhibitors (oxatomide), and trace elements (zinc, iron, copper, selenium, magnesium) [84].
2.6. Role of Antioxidants on Lipid Metabolism during HIV Infection
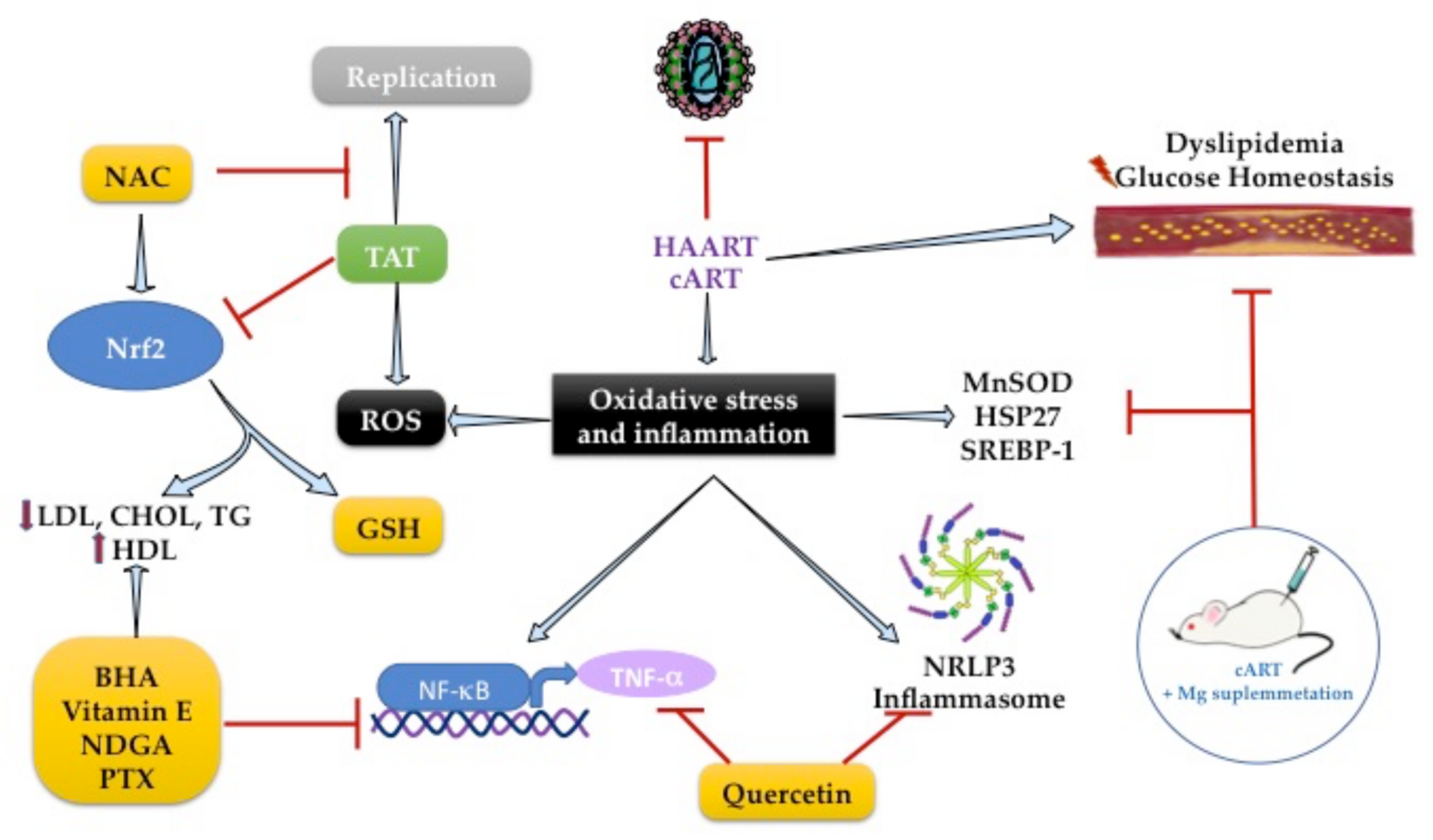
The pathogenesis of dyslipidemia in HIV patients remains poorly understood. Molecular mechanisms are focused on the increased inflammation pathways, and it is known that HAART led to lipid abnormalities such as dyslipidemia. By investigating genes involved in lipid and glucose metabolism alterations in HIV-infected patients treated with HAART, the transcriptional profiling showed an overexpression of MnSOD and heat shock protein (HSP27) in patients with MetS and HAART [85]. That suggests the activation of an endogenous response to the deleterious effects of HAART (Figure 3).
Figure 3. Gene targets antioxidants with antilipidemic effects to reduce oxidative stress and inflammation in HIV infection.
The chronic use of cART with two nucleoside analog inhibitors-NRTIs increases OS and hyperlipidemia caused by lipodystrophy [86]. In HIV-1 transgenic rats, the accumulation of total cholesterol in the liver and serum hypertriglyceridemia is concomitant with higher expression of SREBP-1 in animals treated with cART. However, the enhanced expression of SREBP-1 was suppressed by Mg-supplementation, which was associated with the reduction in serum cholesterol and triglyceride levels [87].
Antioxidants are well known to modulate transcription factors to exert antioxidant and anti-inflammatory effects. The NF-κB/TNF-α pathway was the first inflammatory pathway identified in people with AIDS [88][89]. The use of chain-breaking, lipid-soluble, phenolic antioxidants, such as butylated hydroxyanisole (BHA), ordihydroguaiaretic acid (NDGA), or alpha-tocopherol (vitamin E), can inhibit NF-κB activation because they are peroxyl radical scavengers. BHA also suppresses HIV-enhancing activity through inhibition of NF-κB in lymphoblastoid T-cell and monocytic lymphoblastoid cultures. However, this effect is only achieved if the cells are in an appropriate redox state [88], so the use of compounds that have this duality (antioxidant and anti-inflammatory response) could be a promising therapy.
TNF-cachectin enhances HIV expression and correlates positively with serum T.A.G. levels [89][90], suggesting an excellent pharmacological target for HIV dyslipidemia. In 1993, Dezube et al. showed that pentoxifylline (PTX) reduces TNF-α activity and HIV replication in cultured cells and HIV patients (n = 17). The 8-week study in HIV patients with consumption of PTX (400 mg) reduced TNF-α mRNA and serum T.A.G. levels [89]. PTX is a methylxanthine compound with anti-inflammatory activity inhibiting the NF-κB/TNF-α pathway. It has an antioxidant response by activating the nuclear factor erythroid 2-related factor 2 (NRF2), a transcription factor that regulates the gene expression of endogenous antioxidant enzymes linked to GSH metabolism [91][92].
NRF2 plays a vital role in adipogenesis. Its deficiency in the adipose tissue of ob/ob mice leads to MetS with the aggravation of insulin resistance, hyperglycemia, and hypertriglyceridemia caused by Tat protein, which is one of the six regulatory proteins required for HIV-viral replication [93]. Subsequently, it was described that Tat protein decreased intracellular GSH levels and increased ROS production by enhancing NRF2 expression in MAGI cells. However, using N-acetylcysteine or the overexpression of NRF2 suppressed Tat-induced HIV-1 LTR transactivation, leading to its replicative activity against HIV-1 [94]. This effect was found with the use of tanshinone II-A, a lipid-soluble major monomeric derivative of Salvia miltiorrhiza (Danshen) root that increases GSH levels by regulating NRF2 activation and SIRT1 activity in TZM-bl cells (Figure 3) [95]. Therefore, adequate regulation of NRF2 activity is necessary to maintain intracellular redox homeostasis during harmful signals by HIV infection.
References
- Lot, F.; Cazein, F. Épidémiologie Du VIH et Situation Chez Les Seniors. Soins 2019, 64, 20–24.
- INSIGHT START Study Group. Initiation of Antiretroviral Therapy in Early Asymptomatic HIV Infection. N. Engl. J. Med. 2015, 373, 795–807.
- Taylor, B.S.; Wilkin, T.J.; Shalev, N.; Hammer, S.M. CROI 2013: Advances in Antiretroviral Therapy. Top. Antivir. Med. 2013, 21, 75–89.
- Blas-Garcia, A.; Apostolova, N.; Esplugues, J.V. Oxidative Stress and Mitochondrial Impairment After Treatment with Anti-HIV Drugs: Clinical Implications. Curr. Pharm. Des. 2011, 17, 4076–4086.
- Ivanov, A.V.; Valuev-Elliston, V.T.; Ivanova, O.N.; Kochetkov, S.N.; Starodubova, E.S.; Bartosch, B.; Isaguliants, M.G. Oxidative Stress during HIV Infection: Mechanisms and Consequences. Oxid. Med. Cell. Longev. 2016, 2016, 8910396.
- Chettimada, S.; Lorenz, D.R.; Misra, V.; Dillon, S.T.; Reeves, R.K.; Manickam, C.; Morgello, S.; Kirk, G.D.; Mehta, S.H.; Gabuzda, D. Exosome Markers Associated with Immune Activation and Oxidative Stress in HIV Patients on Antiretroviral Therapy. Sci. Rep. 2018, 8, 7227.
- Sies, H. Oxidative Stress: A Concept in Redox Biology and Medicine. Redox Biol. 2015, 4, 180–183.
- Senoner, T.; Dichtl, W. Oxidative Stress in Cardiovascular Diseases: Still a Therapeutic Target? Nutrients 2019, 11, 2090.
- Ramasamy, I. Update on the Molecular Biology of Dyslipidemias. Clin. Chim. Acta 2016, 454, 143–185.
- Vázquez-Jiménez, J.G.; Roura-Guiberna, A.; Jiménez-Mena, L.R.; Olivares-Reyes, J.A. El Papel de Los Ácidos Grasos Libres En La Resistencia a La Insulina. Gac. Med. Mex. 2017, 153, 852–863.
- Zioga, E.A.M.; Arias-de la Torre, J.; Patera, E.; Borjabad, B.; Macorigh, L.; Ferrer, L. El Papel de Las Intervenciones Biomédicas En La Prevención Del VIH: La Profilaxis Preexposición (PrEP). Med. Fam. SEMERGEN 2020, 46, 202–207.
- Musisi, E.; Matovu, D.K.; Bukenya, A.; Kaswabuli, S.; Zawedde, J.; Andama, A.; Byanyima, P.; Sanyu, I.; Sessolo, A.; Seremba, E.; et al. Effect of Anti-Retroviral Therapy on Oxidative Stress in Hospitalized HIV-Infected Adults with and without TB. Afr. Health Sci. 2018, 18, 512–522.
- García-Fernández, L.; Fiestas, F.; Vásquez, R.; Benites, C. Tratamiento Anti-Retroviral Conteniendo Raltegravir En Mujeres Gestantes Con Infección Por VIH: Revisión Sistemática. Rev. Chil. Infectol. 2016, 33, 60–66.
- Gogineni, V.; Schinazi, R.F.; Hamann, M.T. Role of Marine Natural Products in the Genesis of Antiviral Agents. Chem. Rev. 2015, 115, 9655–9706.
- Salehi, B.; Kumar, N.V.A.; Şener, B.; Sharifi-Rad, M.; Kılıç, M.; Mahady, G.B.; Vlaisavljevic, S.; Iriti, M.; Kobarfard, F.; Setzer, W.N.; et al. Medicinal Plants Used in the Treatment of Human Immunodeficiency Virus. Int. J. Mol. Sci. 2018, 19, 1459.
- Kaur, R.; Sharma, P.; Gupta, G.K.; Ntie-Kang, F.; Kumar, D. Structure-Activity-Relationship and Mechanistic Insights for Anti-HIV Natural Products. Molecules 2020, 25, 2070.
- Shaw, G.M.; Hunter, E. HIV Transmission. Cold Spring Harb. Perspect. Med. 2012, 2, a006965.
- Braun, D.L.; Kouyos, R.D.; Balmer, B.; Grube, C.; Weber, R.; Günthard, H.F. Frequency and Spectrum of Unexpected Clinical Manifestations of Primary HIV-1 Infection. Clin. Infect. Dis. 2015, 61, 1013–1021.
- Tomar, R.H. Breaking the Asymptomatic Phase of HIV-1 Infection. J. Clin. Lab. Anal. 1994, 8, 116–119.
- Moir, S.; Chun, T.-W.; Fauci, A.S. Pathogenic Mechanisms of HIV Disease. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 223–248.
- Min, K.W. Pathology of Acquired Immunodeficiency Syndrome. J. Korean Med. Sci. 1997, 12, 171–184.
- García, M.; Yebra, M.; Vargas, J.A.; Villarreal, M.; Salas, C. Hemophagocytic syndrome associated with lymphoma in an HIV-infected patient: Presentation of a case and review of the literature. Enferm. Infecc. Microbiol. Clin. 1999, 17, 367–368.
- Uldrick, T.S.; Little, R.F. How I Treat Classical Hodgkin Lymphoma in Patients Infected with Human Immunodeficiency Virus. Blood 2015, 125, 1226–1355.
- Magnabosco, G.T.; Lopes, L.M.; Andrade, R.L.d.P.; Brunello, M.E.F.; Monroe, A.A.; Villa, T.C.S. Tuberculosis Control in People Living with HIV/AIDS. Rev. Lat. Am. Enferm. 2016, 24, e2798.
- Gonçalves, P.H.; Uldrick, T.S.; Yarchoan, R. HIV-Associated Kaposi Sarcoma and Related Diseases. AIDS 2017, 31, 1903–1916.
- Kfutwah, A.K.W.; Ngoupo, P.A.T.; Sofeu, C.L.; Ndongo, F.A.; Guemkam, G.; Ndiang, S.T.; Owona, F.; Penda, I.C.; Tchendjou, P.; Rouzioux, C.; et al. Cytomegalovirus Infection in HIV-Infected versus Non-Infected Infants and HIV Disease Progression in Cytomegalovirus Infected versus Non Infected Infants Early Treated with CART in the ANRS 12140-Pediacam Study in Cameroon. BMC Infect. Dis. 2017, 17, 224.
- Castillo-Martínez, N.A.; Mouriño-Pérez, R.R.; Cornejo-Bravo, J.M.; Gaitán-Cepeda, L.A. Factores Relacionados a Candidiasis Oral En Niños y Adolescentes Con VIH, Caracterización de Especies y Susceptibilidad Antifúngica. Rev. Chil. Infectol. 2018, 35, 377–385.
- Mascort, J.; Aguado, C.; Alastrue, I.; Carrillo, R.; Fransi, L.; Zarco, J. HIV and primary care. Think back to AIDS. Aten. Primaria 2017, 49, 65–66.
- Mateo-Urdiales, A.; Johnson, S.; Smith, R.; Nachega, J.B.; Eshun-Wilson, I. Rapid Initiation of Antiretroviral Therapy for People Living with HIV. Cochrane Database Syst. Rev. 2019, 6, CD012962.
- Llibre, J.M.; Fuster-Ruizdeapodaca, M.J.; Rivero, A.; Fernández, E. Cuidados Clínicos Del Paciente Con VIH. Enferm. Infecc. Microbiol. Clin. 2018, 36, 40–44.
- Abuogi, L.L.; Smith, C.; McFarland, E.J. Retention of HIV-Infected Children in the First 12 Months of Anti-Retroviral Therapy and Predictors of Attrition in Resource Limited Settings: A Systematic Review. PLoS ONE 2016, 11, e0156506.
- Pau, A.K.; George, J.M. Antiretroviral Therapy: Current Drugs. Infect. Dis. Clin. N. Am. 2014, 28, 371–402.
- Duracinsky, M.; Leclercq, P.; Armstrong, A.R.; Dolivo, M.; Mouly, F.; Chassany, O. A Longitudinal Evaluation of the Impact of a Polylactic Acid Injection Therapy on Health Related Quality of Life amongst HIV Patients Treated with Anti-Retroviral Agents under Real Conditions of Use. BMC Infect. Dis. 2013, 13, 92.
- Bednasz, C.; Luque, A.E.; Zingman, B.S.; Fischl, M.A.; Gripshover, B.M.; Venuto, C.S.; Gu, J.; Feng, Z.; DiFrancesco, R.; Morse, G.D.; et al. Lipid-Lowering Therapy in HIV-Infected Patients: Relationship with Antiretroviral Agents and Impact of Substance-Related Disorders. Curr. Vasc. Pharmacol. 2016, 14, 280–287.
- Bakari, A.G.; Sani-Bello, F.; Shehu, M.S.; Mai, A.; Aliyu, I.S.; Lawal, I.I. Anti-Retroviral Therapy Induced Diabetes in a Nigerian. Afr. Health Sci. 2007, 7, 133–135.
- Luetkemeyer, A.F.; Havlir, D.V.; Currier, J.S. Complications of HIV Disease and Antiretroviral Treatment. Top. HIV Med. Publ. Int. AIDS Soc. USA 2010, 18, 57–65.
- Katsiki, N.; Mikhailidis, D.P.; Mantzoros, C.S. Non-Alcoholic Fatty Liver Disease and Dyslipidemia: An Update. Metab. Clin. Exp. 2016, 65, 1109–1123.
- Jabarpour, M.; Rashtchizadeh, N.; Argani, H.; Ghorbanihaghjo, A.; Ranjbarzadhag, M.; Sanajou, D.; Panah, F.; Alirezaei, A. The Impact of Dyslipidemia and Oxidative Stress on Vasoactive Mediators in Patients with Renal Dysfunction. Int. Urol. Nephrol. 2019, 51, 2235–2242.
- Black, L.L.; Srivastava, R.; Schoeb, T.R.; Moore, R.D.; Barnes, S.; Kabarowski, J.H. Cholesterol-Independent Suppression of Lymphocyte Activation, Autoimmunity, and Glomerulonephritis by Apolipoprotein A-I in Normocholesterolemic Lupus-Prone Mice. J. Immunol. 2015, 195, 4685–4698.
- Kelesidis, T.; Jackson, N.; McComsey, G.A.; Wang, X.; Elashoff, D.; Dube, M.P.; Brown, T.T.; Yang, O.O.; Stein, J.H.; Currier, J.S. Oxidized Lipoproteins Are Associated with Markers of Inflammation and Immune Activation in HIV-1 Infection. AIDS 2016, 30, 2625–2633.
- Funderburg, N.T.; Mehta, N.N. Lipid Abnormalities and Inflammation in HIV Inflection. Curr. HIV/AIDS Rep. 2016, 13, 218–225.
- Duprez, D.A.; Kuller, L.H.; Tracy, R.; Otvos, J.; Cooper, D.A.; Hoy, J.; Neuhaus, J.; Paton, N.I.; Friis-Moller, N.; Lampe, F.; et al. Lipoprotein Particle Subclasses, Cardiovascular Disease and HIV Infection. Atherosclerosis 2009, 207, 524–529.
- Hussain, I.; Garg, A. Lipodystrophy Syndromes. Endocrinol. Metab. Clin. N. Am. 2016, 45, 783–797.
- Brayson, D.; Frustaci, A.; Verardo, R.; Chimenti, C.; Russo, M.A.; Hayward, R.; Ahmad, S.; Vizcay-Barrena, G.; Protti, A.; Zammit, P.S.; et al. Prelamin A Mediates Myocardial Inflammation in Dilated and HIV-Associated Cardiomyopathies. JCI Insight 2019, 4, e126315.
- Gardner, K.; Hall, P.A.; Chinnery, P.F.; Payne, B.A.I. HIV Treatment and Associated Mitochondrial Pathology: Review of 25 Years of in Vitro, Animal, and Human Studies. Toxicol. Pathol. 2014, 42, 811–822.
- Riddler, S.A.; Smit, E.; Cole, S.R.; Li, R.; Chmiel, J.S.; Dobs, A.; Palella, F.; Visscher, B.; Evans, R.; Kingsley, L.A. Impact of HIV Infection and HAART on Serum Lipids in Men. JAMA 2003, 289, 2978–2982.
- Feeney, E.R.; McAuley, N.; O’Halloran, J.A.; Rock, C.; Low, J.; Satchell, C.S.; Lambert, J.S.; Sheehan, G.J.; Mallon, P.W.G. The Expression of Cholesterol Metabolism Genes in Monocytes from HIV-Infected Subjects Suggests Intracellular Cholesterol Accumulation. J. Infect. Dis. 2013, 207, 628–637.
- Peng, K.; Liu, L.; Wei, D.; Lv, Y.; Wang, G.; Xiong, W.; Wang, X.; Altaf, A.; Wang, L.; He, D.; et al. P2X7R Is Involved in the Progression of Atherosclerosis by Promoting NLRP3 Inflammasome Activation. Int. J. Mol. Med. 2015, 35, 1179–1188.
- Shi, J.; Zhang, Z.; Wu, J. Research Progress on the Relationship between the NLRP3 Inflammasome and Immune Reconstitution in HIV-Infected Patients Receiving Antiretroviral Therapy. Comput. Math. Methods Med. 2022, 2022, 3179200.
- Chivero, E.T.; Guo, M.-L.; Periyasamy, P.; Liao, K.; Callen, S.E.; Buch, S. HIV-1 Tat Primes and Activates Microglial NLRP3 Inflammasome-Mediated Neuroinflammation. J. Neurosci. 2017, 37, 3599–3609.
- Zhang, C.; Song, J.-W.; Huang, H.-H.; Fan, X.; Huang, L.; Deng, J.-N.; Tu, B.; Wang, K.; Li, J.; Zhou, M.-J.; et al. NLRP3 Inflammasome Induces CD4+ T Cell Loss in Chronically HIV-1-Infected Patients. J. Clin. Investig. 2021, 131, e138861.
- Zhang, Y.; Li, X.; Pitzer, A.L.; Chen, Y.; Wang, L.; Li, P.-L. Coronary Endothelial Dysfunction Induced by Nucleotide Oligomerization Domain-like Receptor Protein with Pyrin Domain Containing 3 Inflammasome Activation during Hypercholesterolemia: Beyond Inflammation. Antioxid. Redox Signal. 2015, 22, 1084–1096.
- Vickers, K.C.; Rader, D.J. Nuclear Receptors and MicroRNA-144 Coordinately Regulate Cholesterol Efflux. Circ. Res. 2013, 112, 1529–1531.
- Mujawar, Z.; Rose, H.; Morrow, M.P.; Pushkarsky, T.; Dubrovsky, L.; Mukhamedova, N.; Fu, Y.; Dart, A.; Orenstein, J.M.; Bobryshev, Y.V.; et al. Human Immunodeficiency Virus Impairs Reverse Cholesterol Transport from Macrophages. PLoS Biol. 2006, 4, e365.
- Sopeyin, A.; Zhou, L.; Li, M.; Barakat, L.; Paintsil, E. Dysregulation of Sterol Regulatory Element-Binding Protein 2 Gene in HIV Treatment-Experienced Individuals. PLoS ONE 2019, 14, e0226573.
- Liu, S.-Y.; Aliyari, R.; Chikere, K.; Li, G.; Marsden, M.D.; Smith, J.K.; Pernet, O.; Guo, H.; Nusbaum, R.; Zack, J.A.; et al. Interferon-Inducible Cholesterol-25-Hydroxylase Broadly Inhibits Viral Entry by Production of 25-Hydroxycholesterol. Immunity 2013, 38, 92–105.
- Saulle, I.; Ibba, S.V.; Vittori, C.; Fenizia, C.; Mercurio, V.; Vichi, F.; Caputo, S.L.; Trabattoni, D.; Clerici, M.; Biasin, M. Sterol Metabolism Modulates Susceptibility to HIV-1 Infection. AIDS 2020, 34, 1593–1602.
- Sviridov, D.; Mukhamedova, N.; Makarov, A.A.; Adzhubei, A.; Bukrinsky, M. Comorbidities of HIV Infection: Role of Nef-Induced Impairment of Cholesterol Metabolism and Lipid Raft Functionality. AIDS 2020, 34, 1–13.
- Shrivastav, S.; Kino, T.; Cunningham, T.; Ichijo, T.; Schubert, U.; Heinklein, P.; Chrousos, G.P.; Kopp, J.B. Human Immunodeficiency Virus (HIV)-1 Viral Protein R Suppresses Transcriptional Activity of Peroxisome Proliferator-Activated Receptor and Inhibits Adipocyte Differentiation: Implications for HIV-Associated Lipodystrophy. Mol. Endocrinol. 2008, 22, 234–247.
- Thangavel, S.; Mulet, C.T.; Atluri, V.S.R.; Agudelo, M.; Rosenberg, R.; Devieux, J.G.; Nair, M.P.N. Oxidative Stress in HIV Infection and Alcohol Use: Role of Redox Signals in Modulation of Lipid Rafts and ATP-Binding Cassette Transporters. Antioxid. Redox Signal. 2018, 28, 324–337.
- Arauz, J.; Ramos-Tovar, E.; Muriel, P. Redox State and Methods to Evaluate Oxidative Stress in Liver Damage: From Bench to Bedside. Ann. Hepatol. 2016, 15, 160–173.
- Cadenas, E.; Davies, K.J.A. Mitochondrial Free Radical Generation, Oxidative Stress, and Aging11This Article Is Dedicated to the Memory of Our Dear Friend, Colleague, and Mentor Lars Ernster (1920–1998), in Gratitude for All He Gave to Us. Free Radic. Biol. Med. 2000, 29, 222–230.
- Finkel, T.; Holbrook, N.J. Oxidants, Oxidative Stress and the Biology of Ageing. Nature 2000, 408, 239–247.
- Dröge, W. Free Radicals in the Physiological Control of Cell Function. Physiol. Rev. 2002, 82, 47–95.
- Darley-Usmar, V.; Halliwell, B. Blood Radicals: Reactive Nitrogen Species, Reactive Oxygen Species, Transition Metal Ions, and the Vascular System. Pharm. Res. 1996, 13, 649–662.
- Feillet-Coudray, C.; Fouret, G.; Vigor, C.; Bonafos, B.; Jover, B.; Blachnio-Zabielska, A.; Rieusset, J.; Casas, F.; Gaillet, S.; Landrier, J.F.; et al. Long-Term Measures of Dyslipidemia, Inflammation, and Oxidative Stress in Rats Fed a High-Fat/High-Fructose Diet. Lipids 2019, 54, 81–97.
- Klaunig, E.J. Oxidative Stress and Cancer. Curr. Pharm. Des. 2018, 24, 4771–4778.
- Moldogazieva, N.T.; Mokhosoev, I.M.; Mel’nikova, T.I.; Porozov, Y.B.; Terentiev, A.A. Oxidative Stress and Advanced Lipoxidation and Glycation End Products (ALEs and AGEs) in Aging and Age-Related Diseases. Oxid. Med. Cell. Longev. 2019, 2019, 3085756.
- Mandas, A.; Iorio, E.L.; Congiu, M.G.; Balestrieri, C.; Mereu, A.; Cau, D.; Dessì, S.; Curreli, N. Oxidative Imbalance in HIV-1 Infected Patients Treated with Antiretroviral Therapy. J. Biomed. Biotechnol. 2009, 2009, 749575.
- da Silva, A.S.; Carvalho, T.L.; do Ó, K.P.; da Nóbrega, D.N.; dos Santos Souza, R.; da Silva Lima, V.F.; Farias, I.C.C.; de Mendonça Belmont, T.F.; de Mendonça Cavalcanti, M.d.S.; de Barros Miranda-Filho, D. Association of the Polymorphisms of the Genes APOC3 (Rs2854116), ESR2 (Rs3020450), HFE (Rs1799945), MMP1 (Rs1799750) and PPARG (Rs1801282) with Lipodystrophy in People Living with HIV on Antiretroviral Therapy: A Systematic Review. Mol. Biol. Rep. 2020, 47, 4779–4787.
- Renga, B.; Francisci, D.; Carino, A.; Marchianò, S.; Cipriani, S.; Chiara Monti, M.; Del Sordo, R.; Schiaroli, E.; Distrutti, E.; Baldelli, F.; et al. The HIV Matrix Protein P17 Induces Hepatic Lipid Accumulation via Modulation of Nuclear Receptor Transcriptoma. Sci. Rep. 2015, 5, 15403.
- Kinoo, S.M.; Chuturgoon, A.A.; Singh, B.; Nagiah, S. Hepatic Expression of Cholesterol Regulating Genes Favour Increased Circulating Low-Density Lipoprotein in HIV Infected Patients with Gallstone Disease: A Preliminary Study. BMC Infect. Dis. 2021, 21, 294.
- Guzik, T.J.; West, N.E.J.; Black, E.; McDonald, D.; Ratnatunga, C.; Pillai, R.; Channon, K.M. Vascular Superoxide Production by NAD(P)H Oxidase. Circ. Res. 2000, 86, e85–e90.
- Stokes, K.Y.; Russell, J.M.; Jennings, M.H.; Alexander, J.S.; Granger, D.N. Platelet-Associated NAD(P)H Oxidase Contributes to the Thrombogenic Phenotype Induced by Hypercholesterolemia. Free Radic. Biol. Med. 2007, 43, 22–30.
- Meza, C.A.; La Favor, J.D.; Kim, D.-H.; Hickner, R.C. Endothelial Dysfunction: Is There a Hyperglycemia-Induced Imbalance of NOX and NOS? Int. J. Mol. Sci. 2019, 20, 3775.
- Naito, Y.; Yoshikawa, T. Antioxidants as anti-ageing medicine. Nihon Rinsho Jpn. J. Clin. Med. 2009, 67, 1377–1383.
- Berger, R.G.; Lunkenbein, S.; Ströhle, A.; Hahn, A. Antioxidants in Food: Mere Myth or Magic Medicine? Crit. Rev. Food Sci. Nutr. 2012, 52, 162–171.
- Candas, D.; Li, J.J. MnSOD in Oxidative Stress Response-Potential Regulation via Mitochondrial Protein Influx. Antioxid. Redox Signal. 2014, 20, 1599–1617.
- Valentine, J.S.; Doucette, P.A.; Zittin Potter, S. Copper-Zinc Superoxide Dismutase and Amyotrophic Lateral Sclerosis. Annu. Rev. Biochem. 2005, 74, 563–593.
- Qin, Z.; Reszka, K.J.; Fukai, T.; Weintraub, N.L. Extracellular Superoxide Dismutase (EcSOD) in Vascular Biology: An Update on Exogenous Gene Transfer and Endogenous Regulators of EcSOD. Transl. Res. J. Lab. Clin. Med. 2008, 151, 68–78.
- Olson, K.R.; Gao, Y.; DeLeon, E.R.; Arif, M.; Arif, F.; Arora, N.; Straub, K.D. Catalase as a Sulfide-Sulfur Oxido-Reductase: An Ancient (and Modern?) Regulator of Reactive Sulfur Species (RSS). Redox Biol. 2017, 12, 325–339.
- Hanschmann, E.-M.; Godoy, J.R.; Berndt, C.; Hudemann, C.; Lillig, C.H. Thioredoxins, Glutaredoxins, and Peroxiredoxins—Molecular Mechanisms and Health Significance: From Cofactors to Antioxidants to Redox Signaling. Antioxid. Redox Signal. 2013, 19, 1539–1605.
- Carocho, M.; Ferreira, I.C.F.R. A Review on Antioxidants, Prooxidants and Related Controversy: Natural and Synthetic Compounds, Screening and Analysis Methodologies and Future Perspectives. Food Chem. Toxicol. 2013, 51, 15–25.
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553.
- Esposito, V.; Manente, L.; Viglietti, R.; Parrella, G.; Parrella, R.; Gargiulo, M.; Sangiovanni, V.; Perna, A.; Baldi, A.; De Luca, A.; et al. Comparative Transcriptional Profiling in HIV-Infected Patients Using Human Stress Arrays: Clues to Metabolic Syndrome. Vivo 2012, 26, 237–242.
- van Wijk, J.P.H.; Cabezas, M.C. Hypertriglyceridemia, Metabolic Syndrome, and Cardiovascular Disease in HIV-Infected Patients: Effects of Antiretroviral Therapy and Adipose Tissue Distribution. Int. J. Vasc. Med. 2012, 2012, 201027.
- ElZohary, L.; Weglicki, W.B.; Chmielinska, J.J.; Kramer, J.H.; Mak, I.T. Mg-Supplementation Attenuated Lipogenic and Oxidative/Nitrosative Gene Expression Caused by Combination Antiretroviral Therapy (CART) in HIV-1-Transgenic Rats. PLoS ONE 2019, 14, e0210107.
- Israël, N.; Gougerot-Pocidalo, M.A.; Aillet, F.; Virelizier, J.L. Redox Status of Cells Influences Constitutive or Induced NF-Kappa B Translocation and HIV Long Terminal Repeat Activity in Human T and Monocytic Cell Lines. J. Immunol. 1992, 149, 3386.
- Dezube, B.J.; Pardee, A.B.; Chapman, B.; Beckett, L.A.; Korvick, J.A.; Novick, W.J.; Chiurco, J.; Kasdan, P.; Ahlers, C.M.; Ecto, L.T. Pentoxifylline Decreases Tumor Necrosis Factor Expression and Serum Triglycerides in People with AIDS. NIAID AIDS Clinical Trials Group. J. Acquir. Immune Defic. Syndr. 1993, 6, 787–794.
- Munis, J.R.; Richman, D.D.; Kornbluth, R.S. Human Immunodeficiency Virus-1 Infection of Macrophages in Vitro Neither Induces Tumor Necrosis Factor (TNF)/Cachectin Gene Expression nor Alters TNF/Cachectin Induction by Lipopolysaccharide. J. Clin. Investig. 1990, 85, 591–596.
- Jadeja, R.N.; Upadhyay, K.K.; Devkar, R.V.; Khurana, S. Naturally Occurring Nrf2 Activators: Potential in Treatment of Liver Injury. Oxid. Med. Cell. Longev. 2016, 2016, 3453926.
- Ali, F.E.M.; Bakr, A.G.; Abo-youssef, A.M.; Azouz, A.A.; Hemeida, R.A.M. Targeting Keap-1/Nrf-2 Pathway and Cytoglobin as a Potential Protective Mechanism of Diosmin and Pentoxifylline against Cholestatic Liver Cirrhosis. Life Sci. 2018, 207, 50–60.
- Xue, P.; Hou, Y.; Chen, Y.; Yang, B.; Fu, J.; Zheng, H.; Yarborough, K.; Woods, C.G.; Liu, D.; Yamamoto, M.; et al. Adipose Deficiency of Nrf2 in Ob/Ob Mice Results in Severe Metabolic Syndrome. Diabetes 2013, 62, 845–854.
- Zhang, H.-S.; Li, H.-Y.; Zhou, Y.; Wu, M.-R.; Zhou, H.-S. Nrf2 Is Involved in Inhibiting Tat-Induced HIV-1 Long Terminal Repeat Transactivation. Free Radic. Biol. Med. 2009, 47, 261–268.
- Zhang, H.-S.; Chen, X.-Y.; Wu, T.-C.; Zhang, F.-J. Tanshinone II A Inhibits Tat-Induced HIV-1 Transactivation Through Redox-Regulated AMPK/Nampt Pathway. J. Cell. Physiol. 2014, 229, 1193–1201.
More
Information
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
764
Revisions:
2 times
(View History)
Update Date:
01 Jun 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No