 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Chiu Tsun Philip Tang | -- | 3906 | 2022-05-31 10:46:13 | | | |
| 2 | Rita Xu | + 49 word(s) | 3955 | 2022-06-01 03:50:12 | | |
Video Upload Options
Transforming growth factor-β (TGF-β) is a crucial pathogenic mediator of inflammatory diseases. In tissue fibrosis, TGF-β regulates the pathogenic activity of infiltrated immunocytes and promotes extracellular matrix production via de novo myofibroblast generation and kidney cell activation. However, TGF-β is highly pleiotropic in tissue fibrosis, and thus, direct targeting of TGF-β may also block its protective anti-inflammatory effects, resulting in undesirable outcomes. Increasing evidence suggests the involvement of long non-coding RNAs (lncRNAs) in TGF-β-driven tissue fibrosis with a high cell-type and disease specificity, serving as an ideal target for therapeutic development.
1. Introduction
2. TGF-β1 Signaling in Kidney Diseases
2.1. TGF-β1-Associated lncRNAs in Kidney Diseases
2.1.1. lncRNAs in TGF-β1 Induced EMT
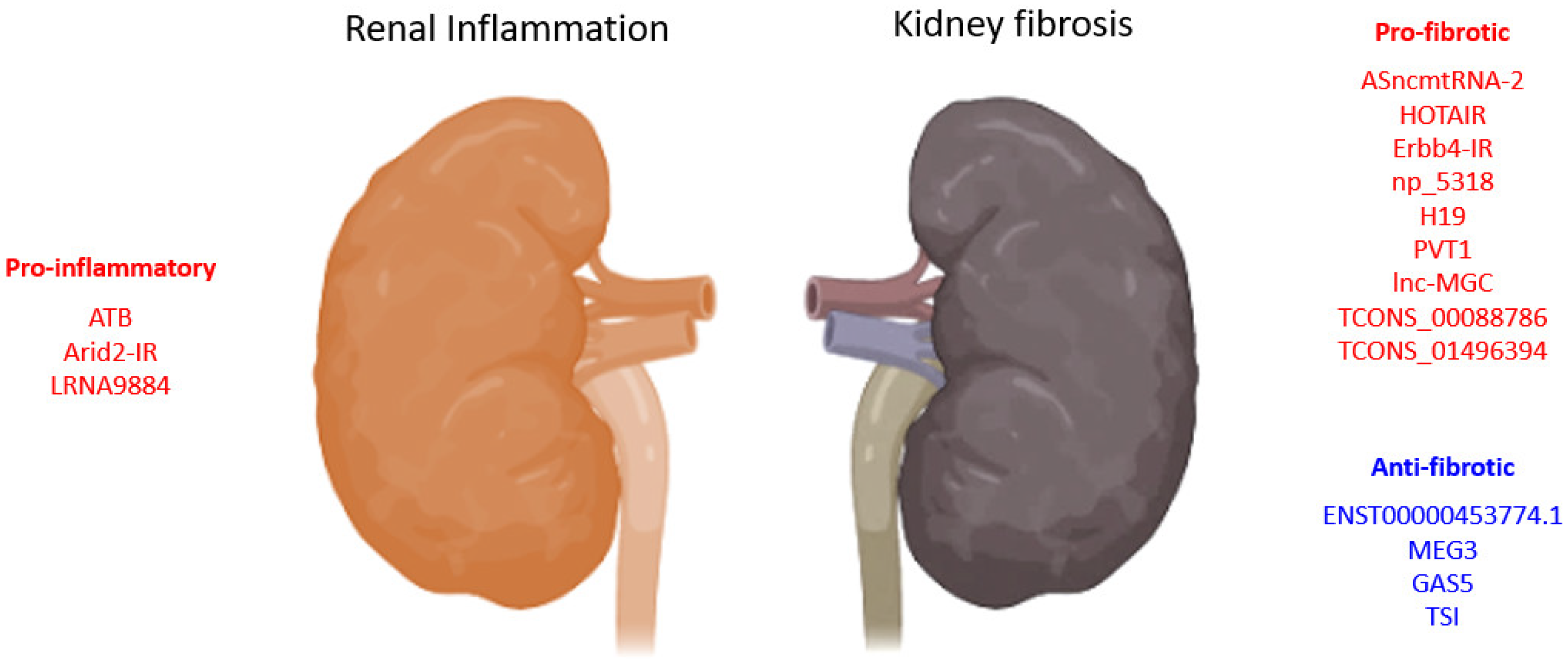
| LncRNA | Biological Process |
Model | Species | Mechanism | Year | Ref. |
|---|---|---|---|---|---|---|
| lnc453774.1 | anti-fibrosis | HK-2 cells | Human | associated with ceRNAs targeting FBN1, IGF1R, KLF7 PPI networks | 2021 | [55] |
| ATB | pro-inflammation | HK-2 cells | Human | promotes apoptosis, senescence, inflammatory cytokines (TNF-α, IL-1β, and IL-6), and adhesion molecules (VCAM-1 and sE-selectin) expression | 2020 | [56] |
| HOTAIR | pro-fibrosis | UUO, TECs-HK-2 |
Human | promotes EMT via Notch1 and miR-124 | 2019 | [52] |
| ENST00000453774.1 | anti-fibrosis | Renal biopsy, UUO, TECs-HK-2 | Human | promotes autophagy (Atg5/7) and Nrf2-driven HO-1 expression and suppresses ECM synthesis (Fn, Col-I) | 2019 | [57] |
| MEG3 | anti-fibrosis | HK-2 cells | Human | suppresses EMT of HK2 cells and is regulated by miR-185/DNMT1/MEG3 pathway | 2019 | [53] |
| TCONS_00088786 | pro-fibrosis | UUO, NRK52E cells | Rat | promotes collagen I, III, and miR-132 expression | 2018 | [54] |
| pro-fibrosis | RNA-seq of rat UUO, NRK52E cells | Rat | promotes Col1a1 and Col3a1 expression | 2017 | [58] | |
| TCONS_01496394 | promotes Ctgf and Fn1 expression | |||||
| ASncmtRNA-2 | pro-fibrosis | HRMC, DN | Human, mouse | promotes TGF-β and Fn1 expression | 2017 | [59] |
| lnc-MGC | pro-fibrosis | STZ-DN, MMC, MCs | Human, mouse | host of miRNA mega-clusters regulating profibrotic genes expression | 2016 | [51] |
| PVT1 | pro-fibrotic | MC, RPTEC, podocytes | Human | PVT1-derived miR-1207-5p-induced TGF-β1, PAI-1, and FN1 | 2013 | [50] |
| pro-fibrotic | ESRD-T2D GWAS | Human | 23 SNPs associated with ESRD | 2007 | [49] |
ceRNAs: competing endogenous RNAs, FBN1: fibrillin-1, IGF1R: insulin-like growth factor 1 receptor, KLF7: Kruppel-like factor 7, PPI: protein-protein interaction, ATB: activated by transforming growth factor-β, TNF-α: tumor necrosis factor alpha, IL: interleukin, VCAM-1: vascular cell adhesion molecule 1, HOTAIR: HOX transcript antisense RNA, UUO: unilateral ureteral obstruction, EMT: epithelial-mesenchymal transition, ECM: extracellular matrix, Fn: fibronectin, Col: collagen, MEG3: maternally expressed gene 3, Ctgf: connective tissue growth factor, ASncmtRNA-2: antisense mitochondrial non-coding RNA-2, HRMC: human renal mesangial cell, DN: diabetic nephropathy, MGC: megacluster, STZ: streptozotocin, MMC: mouse mesangial cell, MCs: mesangial cells, PVT1: plasmacytoma variant translocation 1, RPTEC: human renal proximal tubule epithelial cells, PAI-1: plasminogen activator inhibitor 1, ESRD: end-stage renal disease, T2D: type 2 diabetes, GWAS: genome-wide association studies, SNPs: single nucleotide polymorphisms.
2.1.2. lncRNAs Associated with Reactive Oxygen Species
2.2. Smad3-Dependent lncRNAs in Kidney Diseases
| LncRNA | Biological Process |
Model | Species | Mechanism | Year | Ref. |
|---|---|---|---|---|---|---|
| GAS5 | anti-fibrosis | Smad3-WT/KO UUO, mTECs, MEFs | Mouse | suppresses TGF-β1-induced Col-I/Fn expression and apoptosis, promotes miR-142-5p expression | 2021 | [66] |
| LRNA9884 | pro-inflammation | Cisplatin-AKI, mTECs | Mouse | promotes IL-1β-induced p-p65,TNF-α, MCP-1, and IL-6, binds directly to MIF promoter | 2020 | [69] |
| Smad3-WT/KO-DN, mTECs, SV40 MES 13 | Mouse | Smad3 dependently induced, suppresses IL-1β, TNF-α, and MCP-1, binds directly to the promoter of MCP-1 | 2019 | [67] | ||
| Ptprd-IR (np_4334) | pro-inflammation | mTECs, HEK293T, UUO mice | Human, mouse | Smad3 direct target; promotes inflammatory response and macrophage and T-cell infiltration | 2020 | [70] |
| Erbb4-IR (np_5318) | pro-fibrotic | Smad3-WT/KO-DN, TECs, MCs | Mouse | Smad3 deletion suppressed Erbb4-IR and restored miR-29b expression | 2020 | [71] |
| AKI, PCS-400-012 cells | Human, mouse | promotes I/R-induced renal cell death, further enhances TGF-β1/Smad3 signaling | 2020 | [72] | ||
| UUO, TEC, MEF | Mouse | suppresses Smad7 via promoter binding, enhances Smad3-driven Col-I α-SMA expression | 2018 | [73] | ||
| Smad3-WT/KO-DN, TECs, MCs, MEF | Mouse | enhances Smad3-driven Col-I/IV expression, suppress protective miR-29b via 3’UTR binding | 2018 | [74] | ||
| TSI | anti-fibrosis | UUO, HK2, TECs, MC, HL-7702, LX-2, IMR-90, 16HBE, HKC8 cells | Human, mouse | inhibits Smad3 by direct binding to MH2 domain | 2018 | [68] |
| Arid2-IR | pro-inflammation | UUO, TEC | Mouse | Smad3 direct target; promote fibrotic and inflammatory response, macrophage and T-cell infiltration | 2015 | [75] |
| RNA-seq | pro-fibrotic | UUO /anti-GBM GN of Smad3-WT/KO mice | Mouse | 21 TGF-β/Smad3 dependent lncRNAs | 2014 | [65] |
3. Therapeutic Strategies Targeting lncRNAs
4. Perspectives
References
- Tang, P.M.; Zhang, Y.Y.; Lan, H.Y. LncRNAs in TGF-beta-Driven Tissue Fibrosis. Noncoding RNA 2018, 4, 26.
- Wei, L.H.; Guo, J.U. Coding functions of “noncoding” RNAs. Science 2020, 367, 1074–1075.
- Van der Hauwaert, C.; Glowacki, F.; Pottier, N.; Cauffiez, C. Non-Coding RNAs as New Therapeutic Targets in the Context of Renal Fibrosis. Int. J. Mol. Sci. 2019, 20, 1977.
- Yuan, J.H.; Yang, F.; Wang, F.; Ma, J.Z.; Guo, Y.J.; Tao, Q.F.; Liu, F.; Pan, W.; Wang, T.T.; Zhou, C.C.; et al. A long noncoding RNA activated by TGF-beta promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell 2014, 25, 666–681.
- Winkle, M.; El-Daly, S.M.; Fabbri, M.; Calin, G.A. Noncoding RNA therapeutics—Challenges and potential solutions. Nat. Rev. Drug Discov. 2021, 20, 629–651.
- Chung, J.Y.; Chan, M.K.; Li, J.S.; Chan, A.S.; Tang, P.C.; Leung, K.T.; To, K.F.; Lan, H.Y.; Tang, P.M. TGF-beta Signaling: From Tissue Fibrosis to Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 7575.
- Gil, N.; Ulitsky, I. Regulation of gene expression by cis-acting long non-coding RNAs. Nat. Rev. Genet. 2020, 21, 102–117.
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-beta: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338.
- Tang, P.M.; Nikolic-Paterson, D.J.; Lan, H.Y. Macrophages: Versatile players in renal inflammation and fibrosis. Nat. Rev. Nephrol. 2019, 15, 144–158.
- Zhao, H.; Wu, L.; Yan, G.; Chen, Y.; Zhou, M.; Wu, Y.; Li, Y. Inflammation and tumor progression: Signaling pathways and targeted intervention. Signal Transduct Target. Ther. 2021, 6, 263.
- Meng, X.M.; Tang, P.M.; Li, J.; Lan, H.Y. TGF-beta/Smad signaling in renal fibrosis. Front. Physiol. 2015, 6, 82.
- Tang, P.M.-K.; Lan, H.-Y. MicroRNAs in TGF-β/Smad-mediated Tissue Fibrosis. Curr. Pathobiol. Rep. 2014, 2, 235–243.
- Tang, P.C.; Zhang, Y.Y.; Chan, M.K.; Lam, W.W.; Chung, J.Y.; Kang, W.; To, K.F.; Lan, H.Y.; Tang, P.M. The Emerging Role of Innate Immunity in Chronic Kidney Diseases. Int. J. Mol. Sci. 2020, 21, 4018.
- Colak, S.; Ten Dijke, P. Targeting TGF-beta Signaling in Cancer. Trends Cancer 2017, 3, 56–71.
- Batlle, E.; Massague, J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940.
- Xue, V.W.; Chung, J.Y.; Cordoba, C.A.G.; Cheung, A.H.; Kang, W.; Lam, E.W.; Leung, K.T.; To, K.F.; Lan, H.Y.; Tang, P.M. Transforming Growth Factor-beta: A Multifunctional Regulator of Cancer Immunity. Cancers 2020, 12, 3099.
- Tang, P.M.; Zhang, Y.Y.; Mak, T.S.; Tang, P.C.; Huang, X.R.; Lan, H.Y. Transforming growth factor-beta signalling in renal fibrosis: From Smads to non-coding RNAs. J. Physiol. 2018, 596, 3493–3503.
- Tang, P.M.; Tang, P.C.; Chung, J.Y.; Lan, H.Y. TGF-beta1 signaling in kidney disease: From Smads to long non-coding RNAs. Noncoding RNA Res. 2017, 2, 68–73.
- Lan, H.Y.; Chung, A.C. TGF-beta/Smad signaling in kidney disease. Semin. Nephrol. 2012, 32, 236–243.
- Ma, T.T.; Meng, X.M. TGF-beta/Smad and Renal Fibrosis. Adv. Exp. Med. Biol. 2019, 1165, 347–364.
- Gifford, C.C.; Tang, J.; Costello, A.; Khakoo, N.S.; Nguyen, T.Q.; Goldschmeding, R.; Higgins, P.J.; Samarakoon, R. Negative regulators of TGF-beta1 signaling in renal fibrosis; pathological mechanisms and novel therapeutic opportunities. Clin. Sci. 2021, 135, 275–303.
- Tang, P.C.; Chan, A.S.; Zhang, C.B.; Garcia Cordoba, C.A.; Zhang, Y.Y.; To, K.F.; Leung, K.T.; Lan, H.Y.; Tang, P.M. TGF-beta1 Signaling: Immune Dynamics of Chronic Kidney Diseases. Front. Med. 2021, 8, 628519.
- Robertson, I.B.; Horiguchi, M.; Zilberberg, L.; Dabovic, B.; Hadjiolova, K.; Rifkin, D.B. Latent TGF-beta-binding proteins. Matrix Biol. 2015, 47, 44–53.
- Tang, P.M.; Zhang, Y.Y.; Xiao, J.; Tang, P.C.; Chung, J.Y.; Li, J.; Xue, V.W.; Huang, X.R.; Chong, C.C.; Ng, C.F.; et al. Neural transcription factor Pou4f1 promotes renal fibrosis via macrophage-myofibroblast transition. Proc. Natl. Acad. Sci. USA 2020, 117, 20741–20752.
- Pan, B.; Liu, G.; Jiang, Z.; Zheng, D. Regulation of renal fibrosis by macrophage polarization. Cell Physiol. Biochem. 2015, 35, 1062–1069.
- Lopez-Hernandez, F.J.; Lopez-Novoa, J.M. Role of TGF-beta in chronic kidney disease: An integration of tubular, glomerular and vascular effects. Cell Tissue Res. 2012, 347, 141–154.
- Yang, L.; Pang, Y.; Moses, H.L. TGF-beta and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227.
- Shi, S.; Yu, L.; Zhang, T.; Qi, H.; Xavier, S.; Ju, W.; Bottinger, E. Smad2-dependent downregulation of miR-30 is required for TGF-beta-induced apoptosis in podocytes. PLoS ONE 2013, 8, e75572.
- Herman-Edelstein, M.; Thomas, M.C.; Thallas-Bonke, V.; Saleem, M.; Cooper, M.E.; Kantharidis, P. Dedifferentiation of immortalized human podocytes in response to transforming growth factor-beta: A model for diabetic podocytopathy. Diabetes 2011, 60, 1779–1788.
- Lee, H.S.; Song, C.Y. Differential role of mesangial cells and podocytes in TGF-beta-induced mesangial matrix synthesis in chronic glomerular disease. Histol. Histopathol. 2009, 24, 901–908.
- Sun, Y.B.; Qu, X.; Caruana, G.; Li, J. The origin of renal fibroblasts/myofibroblasts and the signals that trigger fibrosis. Differentiation 2016, 92, 102–107.
- Hinz, B. The extracellular matrix and transforming growth factor-beta1: Tale of a strained relationship. Matrix Biol. 2015, 47, 54–65.
- Lan, H.Y. Tubular epithelial-myofibroblast transdifferentiation mechanisms in proximal tubule cells. Curr. Opin. Nephrol. Hypertens. 2003, 12, 25–29.
- Zhao, Y.; Qiao, X.; Tan, T.K.; Zhao, H.; Zhang, Y.; Liu, L.; Zhang, J.; Wang, L.; Cao, Q.; Wang, Y.; et al. Matrix metalloproteinase 9-dependent Notch signaling contributes to kidney fibrosis through peritubular endothelial-mesenchymal transition. Nephrol. Dial. Transplant. 2017, 32, 781–791.
- Tang, P.M.; Zhou, S.; Li, C.J.; Liao, J.; Xiao, J.; Wang, Q.M.; Lian, G.Y.; Li, J.; Huang, X.R.; To, K.F.; et al. The proto-oncogene tyrosine protein kinase Src is essential for macrophage-myofibroblast transition during renal scarring. Kidney Int. 2018, 93, 173–187.
- Fujimoto, M.; Maezawa, Y.; Yokote, K.; Joh, K.; Kobayashi, K.; Kawamura, H.; Nishimura, M.; Roberts, A.B.; Saito, Y.; Mori, S. Mice lacking Smad3 are protected against streptozotocin-induced diabetic glomerulopathy. Biochem. Biophys. Res. Commun. 2003, 305, 1002–1007.
- Moon, J.A.; Kim, H.T.; Cho, I.S.; Sheen, Y.Y.; Kim, D.K. IN-1130, a novel transforming growth factor-beta type I receptor kinase (ALK5) inhibitor, suppresses renal fibrosis in obstructive nephropathy. Kidney Int. 2006, 70, 1234–1243.
- Sato, M.; Muragaki, Y.; Saika, S.; Roberts, A.B.; Ooshima, A. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J. Clin. Investig. 2003, 112, 1486–1494.
- Sheng, J.; Wang, L.; Tang, P.M.; Wang, H.L.; Li, J.C.; Xu, B.H.; Xue, V.W.; Tan, R.Z.; Jin, N.; Chan, T.F.; et al. Smad3 deficiency promotes beta cell proliferation and function in db/db mice via restoring Pax6 expression. Theranostics 2021, 11, 2845–2859.
- Meng, X.M.; Tang, P.M.; Li, J.; Lan, H.Y. Macrophage Phenotype in Kidney Injury and Repair. Kidney Dis. 2015, 1, 138–146.
- Lv, L.; Tang, P.; You, Y.; Huang, X.; Liu, B.-C.; Lan, H. Long Noncoding RNA-7949 Regulates Macrophage Activation in Renal Inflammation via the TLR4/NF-KB Pathway. Hong Kong J. Nephrol. 2015, 17, S76.
- Wang, S.; Meng, X.M.; Ng, Y.Y.; Ma, F.Y.; Zhou, S.; Zhang, Y.; Yang, C.; Huang, X.R.; Xiao, J.; Wang, Y.Y.; et al. TGF-beta/Smad3 signalling regulates the transition of bone marrow-derived macrophages into myofibroblasts during tissue fibrosis. Oncotarget 2016, 7, 8809–8822.
- Yang, X.; Letterio, J.J.; Lechleider, R.J.; Chen, L.; Hayman, R.; Gu, H.; Roberts, A.B.; Deng, C. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-beta. EMBO J. 1999, 18, 1280–1291.
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914.
- Osielska, M.A.; Jagodzinski, P.P. Long non-coding RNA as potential biomarkers in non-small-cell lung cancer: What do we know so far? Biomed. Pharmacother 2018, 101, 322–333.
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789.
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227.
- Tehrani, S.S.; Ebrahimi, R.; Al, E.A.A.; Panahi, G.; Meshkani, R.; Younesi, S.; Saadat, P.; Parsian, H. Competing Endogenous RNAs (CeRNAs): Novel Network in Neurological Disorders. Curr. Med. Chem. 2021, 28, 5983–6010.
- Hanson, R.L.; Craig, D.W.; Millis, M.P.; Yeatts, K.A.; Kobes, S.; Pearson, J.V.; Lee, A.M.; Knowler, W.C.; Nelson, R.G.; Wolford, J.K. Identification of PVT1 as a candidate gene for end-stage renal disease in type 2 diabetes using a pooling-based genome-wide single nucleotide polymorphism association study. Diabetes 2007, 56, 975–983.
- Alvarez, M.L.; Khosroheidari, M.; Eddy, E.; Kiefer, J. Role of microRNA 1207-5P and its host gene, the long non-coding RNA Pvt1, as mediators of extracellular matrix accumulation in the kidney: Implications for diabetic nephropathy. PLoS ONE 2013, 8, e77468.
- Kato, M.; Wang, M.; Chen, Z.; Bhatt, K.; Oh, H.J.; Lanting, L.; Deshpande, S.; Jia, Y.; Lai, J.Y.; O’Connor, C.L.; et al. An endoplasmic reticulum stress-regulated lncRNA hosting a microRNA megacluster induces early features of diabetic nephropathy. Nat. Commun. 2016, 7, 12864.
- Zhou, H.; Gao, L.; Yu, Z.H.; Hong, S.J.; Zhang, Z.W.; Qiu, Z.Z. LncRNA HOTAIR promotes renal interstitial fibrosis by regulating Notch1 pathway via the modulation of miR-124. Nephrology 2019, 24, 472–480.
- Xue, R.; Li, Y.; Li, X.; Ma, J.; An, C.; Ma, Z. miR-185 affected the EMT, cell viability, and proliferation via DNMT1/MEG3 pathway in TGF-beta1-induced renal fibrosis. Cell Biol. Int. 2019, 43, 1152–1162.
- Zhou, S.G.; Zhang, W.; Ma, H.J.; Guo, Z.Y.; Xu, Y. Silencing of LncRNA TCONS_00088786 reduces renal fibrosis through miR-132. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 166–173.
- Yuan, X.; Tang, W.B.; Peng, L.; Chen, Y.; Tang, S.; Ge, H.; Wang, X.; Xiao, X. Elevation of LncRNA ENST00000453774.1 Prevents Renal Fibrosis by Upregulating FBN1, IGF1R, and KLF7. Kidney Blood Press. Res. 2021, 46, 563–573.
- Sun, H.; Ke, C.; Zhang, L.; Tian, C.; Zhang, Z.; Wu, S. Long Non-Coding RNA (LncRNA)-ATB Promotes Inflammation, Cell Apoptosis and Senescence in Transforming Growth Factor-beta1 (TGF-beta1) Induced Human Kidney 2 (HK-2) Cells via TGFbeta/SMAD2/3 Signaling Pathway. Med. Sci. Monit. 2020, 26, e922029.
- Xiao, X.; Yuan, Q.; Chen, Y.; Huang, Z.; Fang, X.; Zhang, H.; Peng, L.; Xiao, P. LncRNA ENST00000453774.1 contributes to oxidative stress defense dependent on autophagy mediation to reduce extracellular matrix and alleviate renal fibrosis. J. Cell Physiol. 2019, 234, 9130–9143.
- Sun, J.; Zhang, S.; Shi, B.; Zheng, D.; Shi, J. Transcriptome Identified lncRNAs Associated with Renal Fibrosis in UUO Rat Model. Front. Physiol. 2017, 8, 658.
- Gao, Y.; Chen, Z.Y.; Wang, Y.; Liu, Y.; Ma, J.X.; Li, Y.K. Long non-coding RNA ASncmtRNA-2 is upregulated in diabetic kidneys and high glucose-treated mesangial cells. Exp. Ther. Med. 2017, 13, 581–587.
- Irazabal, M.V.; Torres, V.E. Reactive Oxygen Species and Redox Signaling in Chronic Kidney Disease. Cells 2020, 9, 1342.
- Lv, L.L.; Tang, P.M.; Li, C.J.; You, Y.K.; Li, J.; Huang, X.R.; Ni, J.; Feng, M.; Liu, B.C.; Lan, H.Y. The pattern recognition receptor, Mincle, is essential for maintaining the M1 macrophage phenotype in acute renal inflammation. Kidney Int. 2017, 91, 587–602.
- Tang, P.M.; Zhang, Y.Y.; Hung, J.S.; Chung, J.Y.; Huang, X.R.; To, K.F.; Lan, H.Y. DPP4/CD32b/NF-kappaB Circuit: A Novel Druggable Target for Inhibiting CRP-Driven Diabetic Nephropathy. Mol. Ther. 2021, 29, 365–375.
- Sureshbabu, A.; Ryter, S.W.; Choi, M.E. Oxidative stress and autophagy: Crucial modulators of kidney injury. Redox Biol. 2015, 4, 208–214.
- Lai, W.; Tang, Y.; Huang, X.R.; Ming-Kuen Tang, P.; Xu, A.; Szalai, A.J.; Lou, T.Q.; Lan, H.Y. C-reactive protein promotes acute kidney injury via Smad3-dependent inhibition of CDK2/cyclin E. Kidney Int. 2016, 90, 610–626.
- Zhou, Q.; Xiong, Y.; Huang, X.R.; Tang, P.; Yu, X.; Lan, H.Y. Identification of Genes Associated with Smad3-dependent Renal Injury by RNA-seq-based Transcriptome Analysis. Sci. Rep. 2015, 5, 17901.
- Zhang, Y.Y.; Tan, R.Z.; Yu, Y.; Niu, Y.Y.; Yu, C. LncRNA GAS5 protects against TGF-beta-induced renal fibrosis via the Smad3/miRNA-142-5p axis. Am. J. Physiol. Renal. Physiol. 2021, 321, F517–F526.
- Zhang, Y.Y.; Tang, P.M.; Tang, P.C.; Xiao, J.; Huang, X.R.; Yu, C.; Ma, R.C.W.; Lan, H.Y. LRNA9884, a Novel Smad3-Dependent Long Noncoding RNA, Promotes Diabetic Kidney Injury in db/db Mice via Enhancing MCP-1-Dependent Renal Inflammation. Diabetes 2019, 68, 1485–1498.
- Wang, P.; Luo, M.L.; Song, E.; Zhou, Z.; Ma, T.; Wang, J.; Jia, N.; Wang, G.; Nie, S.; Liu, Y.; et al. Long noncoding RNA lnc-TSI inhibits renal fibrogenesis by negatively regulating the TGF-beta/Smad3 pathway. Sci. Transl. Med. 2018, 10, eaat2039.
- Zhang, Y.; Tang, P.M.; Niu, Y.; Garcia Cordoba, C.A.; Huang, X.R.; Yu, C.; Lan, H.Y. Long Non-coding RNA LRNA9884 Promotes Acute Kidney Injury via Regulating NF-kB-Mediated Transcriptional Activation of MIF. Front. Physiol. 2020, 11, 590027.
- Pu, Y.; Zhao, H.; Wu, X.; Mei, M.; Shen, B. The long noncoding RNA Ptprd-IR is a novel molecular target for TGF-beta1-mediated nephritis. Int. J. Biochem. Cell Biol. 2020, 122, 105742.
- Xu, B.H.; Sheng, J.; You, Y.K.; Huang, X.R.; Ma, R.C.W.; Wang, Q.; Lan, H.Y. Deletion of Smad3 prevents renal fibrosis and inflammation in type 2 diabetic nephropathy. Metabolism 2020, 103, 154013.
- Lu, J.; Miao, J.; Sun, J. LncRNA np_5318 promotes renal ischemia-reperfusion injury through the TGF-beta/Smad signaling pathway. Exp. Ther. Med. 2020, 19, 2833–2840.
- Feng, M.; Tang, P.M.; Huang, X.R.; Sun, S.F.; You, Y.K.; Xiao, J.; Lv, L.L.; Xu, A.P.; Lan, H.Y. TGF-beta Mediates Renal Fibrosis via the Smad3-Erbb4-IR Long Noncoding RNA Axis. Mol. Ther. 2018, 26, 148–161.
- Sun, S.F.; Tang, P.M.K.; Feng, M.; Xiao, J.; Huang, X.R.; Li, P.; Ma, R.C.W.; Lan, H.Y. Novel lncRNA Erbb4-IR Promotes Diabetic Kidney Injury in db/db Mice by Targeting miR-29b. Diabetes 2018, 67, 731–744.
- Zhou, Q.; Huang, X.R.; Yu, J.; Yu, X.; Lan, H.Y. Long Noncoding RNA Arid2-IR Is a Novel Therapeutic Target for Renal Inflammation. Mol. Ther. 2015, 23, 1034–1043.
- Bommireddy, R.; Engle, S.J.; Ormsby, I.; Boivin, G.P.; Babcock, G.F.; Doetschman, T. Elimination of both CD4+ and CD8+ T cells but not B cells eliminates inflammation and prolongs the survival of TGFbeta1-deficient mice. Cell Immunol. 2004, 232, 96–104.
- Vincenti, F.; Fervenza, F.C.; Campbell, K.N.; Diaz, M.; Gesualdo, L.; Nelson, P.; Praga, M.; Radhakrishnan, J.; Sellin, L.; Singh, A.; et al. A Phase 2, Double-Blind, Placebo-Controlled, Randomized Study of Fresolimumab in Patients With Steroid-Resistant Primary Focal Segmental Glomerulosclerosis. Kidney Int. Rep. 2017, 2, 800–810.
- Lacouture, M.E.; Morris, J.C.; Lawrence, D.P.; Tan, A.R.; Olencki, T.E.; Shapiro, G.I.; Dezube, B.J.; Berzofsky, J.A.; Hsu, F.J.; Guitart, J. Cutaneous keratoacanthomas/squamous cell carcinomas associated with neutralization of transforming growth factor beta by the monoclonal antibody fresolimumab (GC1008). Cancer Immunol. Immunother. 2015, 64, 437–446.
- Sun, S.F.; Tang, P.M.K.; Huang, X.R.; Lan, H.Y. Response letter: “Novel lncRNA Erbb4-IR promotes diabetic kidney injury in db/db mice by targeting miR-29b”. Transl. Cancer Res. 2018, 67, S629–S631.
- Feng, M.; Tang, P.M.-K.; You, Y.-K.; Lv, L.-L.; Huang, X.-R.; Xu, A.; Lan, H. Long Non-coding RNA_5318 is a Novel Therapeutic Target for Renal Fibrosis in Obstructive Nephropathy. Hong Kong J. Nephrol. 2015, 17, S62–S63.
- Shen, S.; Wang, J.; Zheng, B.; Tao, Y.; Li, M.; Wang, Y.; Ni, X.; Suo, T.; Liu, H.; Liu, H.; et al. LINC01714 Enhances Gemcitabine Sensitivity by Modulating FOXO3 Phosphorylation in Cholangiocarcinoma. Mol. Ther. Nucleic Acids 2020, 19, 446–457.
- Maruyama, R.; Yokota, T. Knocking Down Long Noncoding RNAs Using Antisense Oligonucleotide Gapmers. Methods Mol. Biol. 2020, 2176, 49–56.
- Rinaldi, C.; Wood, M.J.A. Antisense oligonucleotides: The next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 2018, 14, 9–21.
- Lima, W.F.; Vickers, T.A.; Nichols, J.; Li, C.; Crooke, S.T. Defining the factors that contribute to on-target specificity of antisense oligonucleotides. PLoS ONE 2014, 9, e101752.
- Dhuri, K.; Bechtold, C.; Quijano, E.; Pham, H.; Gupta, A.; Vikram, A.; Bahal, R. Antisense Oligonucleotides: An Emerging Area in Drug Discovery and Development. J. Clin. Med. 2020, 9, 2004.
- Zanardi, T.A.; Kim, T.W.; Shen, L.; Serota, D.; Papagiannis, C.; Park, S.Y.; Kim, Y.; Henry, S.P. Chronic Toxicity Assessment of 2′-O-Methoxyethyl Antisense Oligonucleotides in Mice. Nucleic Acid Ther. 2018, 28, 233–241.
- Sennoga, C.A.; Kanbar, E.; Auboire, L.; Dujardin, P.A.; Fouan, D.; Escoffre, J.M.; Bouakaz, A. Microbubble-mediated ultrasound drug-delivery and therapeutic monitoring. Exp. Opin. Drug Deliv. 2017, 14, 1031–1043.
- Xue, V.W.; Chung, J.Y.; Tang, P.C.; Chan, A.S.; To, T.H.; Chung, J.S.; Mussal, F.; Lam, E.W.; Li, C.; To, K.F.; et al. USMB-shMincle: A virus-free gene therapy for blocking M1/M2 polarization of tumor-associated macrophages. Mol. Ther. Oncolytics 2021, 23, 26–37.
- Chen, Q.; Su, Y.; He, X.; Zhao, W.; Wu, C.; Zhang, W.; Si, X.; Dong, B.; Zhao, L.; Gao, Y.; et al. Plasma long non-coding RNA MALAT1 is associated with distant metastasis in patients with epithelial ovarian cancer. Oncol. Lett. 2016, 12, 1361–1366.
- Noviello, T.M.R.; Di Liddo, A.; Ventola, G.M.; Spagnuolo, A.; D’Aniello, S.; Ceccarelli, M.; Cerulo, L. Detection of long non-coding RNA homology, a comparative study on alignment and alignment-free metrics. BMC Bioinform. 2018, 19, 407.
- Xue, W.J.; Ying, X.L.; Jiang, J.H.; Xu, Y.H. Prostate cancer antigen 3 as a biomarker in the urine for prostate cancer diagnosis: A meta-analysis. J. Cancer Res. Ther. 2014, 10, C218–C221.
- Luan, Y.; Li, X.; Luan, Y.; Zhao, R.; Li, Y.; Liu, L.; Hao, Y.; Oleg Vladimir, B.; Jia, L. Circulating lncRNA UCA1 Promotes Malignancy of Colorectal Cancer via the miR-143/MYO6 Axis. Mol. Ther. Nucleic Acids 2020, 19, 790–803.
- Omura, J.; Habbout, K.; Shimauchi, T.; Wu, W.H.; Breuils-Bonnet, S.; Tremblay, E.; Martineau, S.; Nadeau, V.; Gagnon, K.; Mazoyer, F.; et al. Identification of Long Noncoding RNA H19 as a New Biomarker and Therapeutic Target in Right Ventricular Failure in Pulmonary Arterial Hypertension. Circulation 2020, 142, 1464–1484.
- Alfaifi, M.; Ali Beg, M.M.; Alshahrani, M.Y.; Ahmad, I.; Alkhathami, A.G.; Joshi, P.C.; Alshehri, O.M.; Alamri, A.M.; Verma, A.K. Circulating long non-coding RNAs NKILA, NEAT1, MALAT1, and MIAT expression and their association in type 2 diabetes mellitus. BMJ Open Diabetes Res. Care 2021, 9, e001821.


