Transforming growth factor-β (TGF-β) is a crucial pathogenic mediator of inflammatory diseases. In tissue fibrosis, TGF-β regulates the pathogenic activity of infiltrated immunocytes and promotes extracellular matrix production via de novo myofibroblast generation and kidney cell activation. However, TGF-β is highly pleiotropic in tissue fibrosis, and thus, direct targeting of TGF-β may also block its protective anti-inflammatory effects, resulting in undesirable outcomes. Increasing evidence suggests the involvement of long non-coding RNAs (lncRNAs) in TGF-β-driven tissue fibrosis with a high cell-type and disease specificity, serving as an ideal target for therapeutic development.
- long non-coding RNA
- fibrosis
- transforming growth factor-β
1. Introduction
2. TGF-β1 Signaling in Kidney Diseases
2.1. TGF-β1-Associated lncRNAs in Kidney Diseases
2.1.1. lncRNAs in TGF-β1 Induced EMT
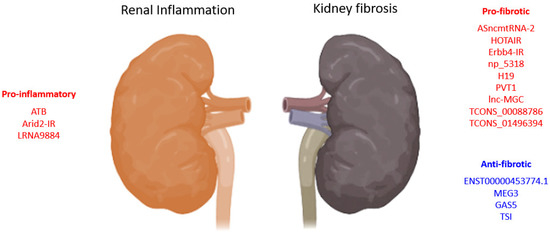
| LncRNA | Biological Process |
Model | Species | Mechanism | Year | Ref. |
|---|---|---|---|---|---|---|
| lnc453774.1 | anti-fibrosis | HK-2 cells | Human | associated with ceRNAs targeting FBN1, IGF1R, KLF7 PPI networks | 2021 | [61] |
| ATB | pro-inflammation | HK-2 cells | Human | promotes apoptosis, senescence, inflammatory cytokines (TNF-α, IL-1β, and IL-6), and adhesion molecules (VCAM-1 and sE-selectin) expression | 2020 | [62] |
| HOTAIR | pro-fibrosis | UUO, TECs-HK-2 |
Human | promotes EMT via Notch1 and miR-124 | 2019 | [58] |
| ENST00000453774.1 | anti-fibrosis | Renal biopsy, UUO, TECs-HK-2 | Human | promotes autophagy (Atg5/7) and Nrf2-driven HO-1 expression and suppresses ECM synthesis (Fn, Col-I) | 2019 | [63] |
| MEG3 | anti-fibrosis | HK-2 cells | Human | suppresses EMT of HK2 cells and is regulated by miR-185/DNMT1/MEG3 pathway | 2019 | [59] |
| TCONS_00088786 | pro-fibrosis | UUO, NRK52E cells | Rat | promotes collagen I, III, and miR-132 expression | 2018 | [60] |
| pro-fibrosis | RNA-seq of rat UUO, NRK52E cells | Rat | promotes Col1a1 and Col3a1 expression | 2017 | [64] | |
| TCONS_01496394 | promotes Ctgf and Fn1 expression | |||||
| ASncmtRNA-2 | pro-fibrosis | HRMC, DN | Human, mouse | promotes TGF-β and Fn1 expression | 2017 | [65] |
| lnc-MGC | pro-fibrosis | STZ-DN, MMC, MCs | Human, mouse | host of miRNA mega-clusters regulating profibrotic genes expression | 2016 | [57] |
| PVT1 | pro-fibrotic | MC, RPTEC, podocytes | Human | PVT1-derived miR-1207-5p-induced TGF-β1, PAI-1, and FN1 | 2013 | [56] |
| pro-fibrotic | ESRD-T2D GWAS | Human | 23 SNPs associated with ESRD | 2007 | [55] |
ceRNAs: competing endogenous RNAs, FBN1: fibrillin-1, IGF1R: insulin-like growth factor 1 receptor, KLF7: Kruppel-like factor 7, PPI: protein-protein interaction, ATB: activated by transforming growth factor-β, TNF-α: tumor necrosis factor alpha, IL: interleukin, VCAM-1: vascular cell adhesion molecule 1, HOTAIR: HOX transcript antisense RNA, UUO: unilateral ureteral obstruction, EMT: epithelial-mesenchymal transition, ECM: extracellular matrix, Fn: fibronectin, Col: collagen, MEG3: maternally expressed gene 3, Ctgf: connective tissue growth factor, ASncmtRNA-2: antisense mitochondrial non-coding RNA-2, HRMC: human renal mesangial cell, DN: diabetic nephropathy, MGC: megacluster, STZ: streptozotocin, MMC: mouse mesangial cell, MCs: mesangial cells, PVT1: plasmacytoma variant translocation 1, RPTEC: human renal proximal tubule epithelial cells, PAI-1: plasminogen activator inhibitor 1, ESRD: end-stage renal disease, T2D: type 2 diabetes, GWAS: genome-wide association studies, SNPs: single nucleotide polymorphisms.
2.1.2. lncRNAs Associated with Reactive Oxygen Species
2.2. Smad3-Dependent lncRNAs in Kidney Diseases
| LncRNA | Biological Process |
Model | Species | Mechanism | Year | Ref. |
|---|---|---|---|---|---|---|
| GAS5 | anti-fibrosis | Smad3-WT/KO UUO, mTECs, MEFs | Mouse | suppresses TGF-β1-induced Col-I/Fn expression and apoptosis, promotes miR-142-5p expression | 2021 | [72] |
| LRNA9884 | pro-inflammation | Cisplatin-AKI, mTECs | Mouse | promotes IL-1β-induced p-p65,TNF-α, MCP-1, and IL-6, binds directly to MIF promoter | 2020 | [75] |
| Smad3-WT/KO-DN, mTECs, SV40 MES 13 | Mouse | Smad3 dependently induced, suppresses IL-1β, TNF-α, and MCP-1, binds directly to the promoter of MCP-1 | 2019 | [73] | ||
| Ptprd-IR (np_4334) | pro-inflammation | mTECs, HEK293T, UUO mice | Human, mouse | Smad3 direct target; promotes inflammatory response and macrophage and T-cell infiltration | 2020 | [76] |
| Erbb4-IR (np_5318) | pro-fibrotic | Smad3-WT/KO-DN, TECs, MCs | Mouse | Smad3 deletion suppressed Erbb4-IR and restored miR-29b expression | 2020 | [77] |
| AKI, PCS-400-012 cells | Human, mouse | promotes I/R-induced renal cell death, further enhances TGF-β1/Smad3 signaling | 2020 | [78] | ||
| UUO, TEC, MEF | Mouse | suppresses Smad7 via promoter binding, enhances Smad3-driven Col-I α-SMA expression | 2018 | [79] | ||
| Smad3-WT/KO-DN, TECs, MCs, MEF | Mouse | enhances Smad3-driven Col-I/IV expression, suppress protective miR-29b via 3’UTR binding | 2018 | [80] | ||
| TSI | anti-fibrosis | UUO, HK2, TECs, MC, HL-7702, LX-2, IMR-90, 16HBE, HKC8 cells | Human, mouse | inhibits Smad3 by direct binding to MH2 domain | 2018 | [74] |
| Arid2-IR | pro-inflammation | UUO, TEC | Mouse | Smad3 direct target; promote fibrotic and inflammatory response, macrophage and T-cell infiltration | 2015 | [81] |
| RNA-seq | pro-fibrotic | UUO /anti-GBM GN of Smad3-WT/KO mice | Mouse | 21 TGF-β/Smad3 dependent lncRNAs | 2014 | [71] |
Smad3-dependent lncRNAs with therapeutic potential in renal diseases have been identified in previous studies. Inhibition of Erbb4-IR alleviated renal fibrosis in fibrotic UUO and DN models [5][6]. Inhibition of Arid2, LncRNA_5318, and LRNA9884 also suppressed renal inflammation in UUO and diabetic models [7][8][9]. LncRNA can be targeted by antisense-based strategies or by shRNAs, consisting of siRNAs and modified antisense oligonucleotides (ASOs) [10]. The ASO-based technologies including novel chemical modifications were optimized with multiple preclinical trials, and the efficiency of cellular uptake and the expression levels of targeted ncRNAs have largely improved[11]. The inhibition efficiency and toxicity were major concerns of directly administering lncRNA-targeting agents via tissue or tail vein injection, where toxicity is observed in a dose-dependent manner, i.e., off-target effects through nonspecific binding to similar nucleotide sequences [12][13][14]. However, repeated high dosages of siRNAs and gapmers are required for effective lncRNA inhibition in vivo. Therefore, post-delivery monitoring and optimizing effective concentration of lncRNAs therapeutics are critical for translational application. Novel noninvasive
ultrasound microbubble-assisted (USMB) delivery largely reduced the concentrations of lncRNA-targeting agents in nontargeted tissue [15], contributing to the safety and effectiveness in preclinical studies[16][5][17]. Thus, USMB represents a realistic approach to translating lncRNA-targeted therapeutics with added value in postdelivery monitoring and assessment with its imaging function. Moreover, among the FDA- (Food and Drug Administration) and EMA- (European Medicines Agency) approved ASO-based therapies targeting mRNA expression in the liver, most were administered subcutaneously (mipomersen, inotersen, givosiran, volanesorsen, inclisiran, and lumasiran)[18], suggesting the potential for developing subcutaneously delivered lncRNA-targeted ASOs for kidney fibrosis.
4. Perspectives
The translational development of therapeutics targeting the TGF-β1 signaling pathway has been largely hindered by its key regulatory roles in multiple physiological processes. In recent decades, the dissection of TGF-β1 signaling pathways has revealed numerous precise therapeutic targets, including lncRNAs for inflammatory diseases. Emerging evidence shows that lncRNAs are specific pathogenic mediators of TGF-β1, regulating a particular function of TGF-β1 during inflammatory disease progression that can be targeted to develop effective gene-based therapies. With the advancement of RNA sequencing at single-cell resolution and bioinformatic analysis, a more in-depth regulatory mechanism of lncRNAs in inflammatory diseases will be discovered. Disease- and cell-type-specific lncRNAs will be identified for the development of precision therapies against tissue inflammation.
This entry is adapted from the peer-reviewed paper 10.3390/ncrna8030036
References
- Hui Y. Lan; Arthur C.-K. Chung; TGF-β/Smad Signaling in Kidney Disease. Seminars in Nephrology 2012, 32, 236-243, 10.1016/j.semnephrol.2012.04.002.
- Ramireddy Bommireddy; Sandra J. Engle; Ilona Ormsby; Gregory P. Boivin; George F. Babcock; Thomas Doetschman; Elimination of both CD4+ and CD8+ T cells but not B cells eliminates inflammation and prolongs the survival of TGFβ1-deficient mice. Cellular Immunology 2004, 232, 96-104, 10.1016/j.cellimm.2005.02.004.
- Flavio Vincenti; Fernando C. Fervenza; Kirk N. Campbell; Montserrat Diaz; Loreto Gesualdo; Peter Nelson; Manuel Praga; Jai Radhakrishnan; Lorenz Sellin; Ajay Singh; et al. A Phase 2, Double-Blind, Placebo-Controlled, Randomized Study of Fresolimumab in Patients With Steroid-Resistant Primary Focal Segmental Glomerulosclerosis. Kidney International Reports 2017, 2, 800-810, 10.1016/j.ekir.2017.03.011.
- Mario E. Lacouture; John C. Morris; Donald P. Lawrence; Antoinette R. Tan; Thomas E. Olencki; Geoffrey I. Shapiro; Bruce J. Dezube; Jay A. Berzofsky; Frank J. Hsu; Joan Guitart; et al. Cutaneous keratoacanthomas/squamous cell carcinomas associated with neutralization of transforming growth factor β by the monoclonal antibody fresolimumab (GC1008). Medical Oncology and Tumor Pharmacotherapy 2015, 64, 437-446, 10.1007/s00262-015-1653-0.
- Si F. Sun; Patrick M.K. Tang; Min Feng; Jun Xiao; Xiao R. Huang; Ping Li; Ronald C.W. Ma; Hui Y. Lan; Novel lncRNA Erbb4-IR Promotes Diabetic Kidney Injury in db/db Mice by Targeting miR-29b. Diabetes 2017, 67, 731-744, 10.2337/db17-0816.
- Min Feng; Patrick Ming Kuen Tang; Xiao-Ru Huang; Si-Fan Sun; Yong-Ke You; Jun Xiao; Lin-Li Lv; An-Ping Xu; Hui-Yao Lan; TGF-β Mediates Renal Fibrosis via the Smad3-Erbb4-IR Long Noncoding RNA Axis. Molecular Therapy 2018, 26, 148-161, 10.1016/j.ymthe.2017.09.024.
- Ying-Ying Zhang; Patrick Ming-Kuen Tang; Philip Chiu-Tsun Tang; Jun Xiao; Xiao-Ru Huang; Chen Yu; Ronald C.W. Ma; Hui-Yao Lan; LRNA9884, a Novel Smad3-Dependent Long Noncoding RNA, Promotes Diabetic Kidney Injury in db/db Mice via Enhancing MCP-1–Dependent Renal Inflammation. Diabetes 2019, 68, 1485-1498, 10.2337/db18-1075.
- Qin Zhou; Xiao R Huang; Jianwen Yu; Xueqing Yu; Hui Y Lan; Long Noncoding RNA Arid2-IR Is a Novel Therapeutic Target for Renal Inflammation. Molecular Therapy 2015, 23, 1034-1043, 10.1038/mt.2015.31.
- Ying-Ying Zhang; Patrick Ming-Kuen Tang; Philip Chiu-Tsun Tang; Jun Xiao; Xiao-Ru Huang; Chen Yu; Ronald C.W. Ma; Hui-Yao Lan; LRNA9884, a Novel Smad3-Dependent Long Noncoding RNA, Promotes Diabetic Kidney Injury in db/db Mice via Enhancing MCP-1–Dependent Renal Inflammation. Diabetes 2019, 68, 1485-1498, 10.2337/db18-1075.
- Rika Maruyama; Toshifumi Yokota; Knocking Down Long Noncoding RNAs Using Antisense Oligonucleotide Gapmers. Cell Engineering 2020, ., 49-56, 10.1007/978-1-0716-0771-8_3.
- Carlo Rinaldi; Matthew J. A. Wood; Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nature Reviews Neurology 2017, 14, 9-21, 10.1038/nrneurol.2017.148.
- Walt F. Lima; Timothy A. Vickers; Josh Nichols; Cheryl Li; Stanley T. Crooke; Defining the Factors That Contribute to On-Target Specificity of Antisense Oligonucleotides. PLoS ONE 2014, 9, e101752, 10.1371/journal.pone.0101752.
- Karishma Dhuri; Clara Bechtold; Elias Quijano; Ha Pham; Anisha Gupta; Ajit Vikram; Raman Bahal; Antisense Oligonucleotides: An Emerging Area in Drug Discovery and Development. Journal of Clinical Medicine 2020, 9, 2004, 10.3390/jcm9062004.
- Thomas A. Zanardi; Tae-Won Kim; Lijiang Shen; David Serota; Chris Papagiannis; Shin-Young Park; Yunlip Kim; Scott P. Henry; Chronic Toxicity Assessment of 2′-O-Methoxyethyl Antisense Oligonucleotides in Mice. Nucleic Acid Therapeutics 2018, 28, 233-241, 10.1089/nat.2017.0706.
- Charles A. Sennoga; Emma Kanbar; Laurent Auboire; Paul-Armand Dujardin; Damien Fouan; Jean-Michel Escoffre; Ayache Bouakaz; Microbubble-mediated ultrasound drug-delivery and therapeutic monitoring. Expert Opinion on Drug Delivery 2016, 14, 1031-1043, 10.1080/17425247.2017.1266328.
- Ying-Ying Zhang; Patrick Ming-Kuen Tang; Philip Chiu-Tsun Tang; Jun Xiao; Xiao-Ru Huang; Chen Yu; Ronald C.W. Ma; Hui-Yao Lan; LRNA9884, a Novel Smad3-Dependent Long Noncoding RNA, Promotes Diabetic Kidney Injury in db/db Mice via Enhancing MCP-1–Dependent Renal Inflammation. Diabetes 2019, 68, 1485-1498, 10.2337/db18-1075.
- Vivian Weiwen Xue; Jeff Yat-Fai Chung; Philip Chiu-Tsun Tang; Alex Siu-Wing Chan; Travis Hoi-Wai To; Justin Shing-Yin Chung; Francis Mussal; Eric W.-F. Lam; Chunjie Li; Ka-Fai To; et al. USMB-shMincle: a virus-free gene therapy for blocking M1/M2 polarization of tumor-associated macrophages. Molecular Therapy - Oncolytics 2021, 23, 26-37, 10.1016/j.omto.2021.08.010.
- Melanie Winkle; Sherien M. El-Daly; Muller Fabbri; George A. Calin; Noncoding RNA therapeutics — challenges and potential solutions. Nature Reviews Drug Discovery 2021, 20, 629-651, 10.1038/s41573-021-00219-z.
- Qingjuan Chen; Yongyong Su; Xiaopeng He; Weian Zhao; Caixia Wu; Weibo Zhang; Xiaomin Si; Bingwei Dong; Lianying Zhao; Yufang Gao; et al. Plasma long non-coding RNA MALAT1 is associated with distant metastasis in patients with epithelial ovarian cancer. Oncology Letters 2016, 12, 1361-1366, 10.3892/ol.2016.4800.
- Teresa M. R. Noviello; Antonella Di Liddo; Giovanna M. M. Ventola; Antonietta Spagnuolo; Salvatore D’Aniello; Michele Ceccarelli; Luigi Cerulo; Detection of long non–coding RNA homology, a comparative study on alignment and alignment–free metrics. BMC Bioinformatics 2018, 19, 1-12, 10.1186/s12859-018-2441-6.
- Ye-Hui Xu; Wu-Jin Xue; Xiu-Li Ying; Jin-Hong Jiang; Prostate cancer antigen 3 as a biomarker in the urine for prostate cancer diagnosis: A meta-analysis. Journal of Cancer Research and Therapeutics 2014, 10, 218, 10.4103/0973-1482.145881.
- Yunpeng Luan; Xiang Li; Yunqi Luan; Rong Zhao; Yanmei Li; Lili Liu; Yizhuo Hao; Burakovaov Oleg Vladimir; Lu Jia; Circulating lncRNA UCA1 Promotes Malignancy of Colorectal Cancer via the miR-143/MYO6 Axis. Molecular Therapy - Nucleic Acids 2019, 19, 790-803, 10.1016/j.omtn.2019.12.009.
- Junichi Omura; Karima Habbout; Tsukasa Shimauchi; Wen-Hui Wu; Sandra Breuils-Bonnet; Eve Tremblay; Sandra Martineau; Valérie Nadeau; Kassandra Gagnon; Florence Mazoyer; et al. Identification of Long Noncoding RNA H19 as a New Biomarker and Therapeutic Target in Right Ventricular Failure in Pulmonary Arterial Hypertension. Circulation 2020, 142, 1464-1484, 10.1161/circulationaha.120.047626.
- Mohammed Alfaifi; Mirza Masroor Ali Beg; Mohammad Yahya Alshahrani; Irfan Ahmad; Ali Gaithan Alkhathami; Prakash C Joshi; Osama M Alshehri; Abdulrahman Manaa Alamri; Amit Kumar Verma; Circulating long non-coding RNAs NKILA, NEAT1, MALAT1, and MIAT expression and their association in type 2 diabetes mellitus. BMJ Open Diabetes Research & Care 2021, 9, e001821, 10.1136/bmjdrc-2020-001821.