Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mohammad Islam | -- | 2520 | 2022-05-30 12:31:35 | | | |
| 2 | Beatrix Zheng | + 1 word(s) | 2521 | 2022-05-31 03:35:22 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Islam, M.; Alzawi, A.; , .; Ellis, I. Targeting Akt in Treating Head and Neck Cancer. Encyclopedia. Available online: https://encyclopedia.pub/entry/23560 (accessed on 30 June 2026).
Islam M, Alzawi A, , Ellis I. Targeting Akt in Treating Head and Neck Cancer. Encyclopedia. Available at: https://encyclopedia.pub/entry/23560. Accessed June 30, 2026.
Islam, Mohammad, Albashir Alzawi, , Ian Ellis. "Targeting Akt in Treating Head and Neck Cancer" Encyclopedia, https://encyclopedia.pub/entry/23560 (accessed June 30, 2026).
Islam, M., Alzawi, A., , ., & Ellis, I. (2022, May 30). Targeting Akt in Treating Head and Neck Cancer. In Encyclopedia. https://encyclopedia.pub/entry/23560
Islam, Mohammad, et al. "Targeting Akt in Treating Head and Neck Cancer." Encyclopedia. Web. 30 May, 2022.
Copy Citation
When Akt, a signalling protein, is activated by different growth factors such as epidermal growth factor, transforming growth factor α/β, vascular endothelial growth factor and nerve growth factor, head and neck cancer cell spreading is stimulated. Tumour microenvironment plays an important role in cancer spreading by synthesising and secreting growth factors and suggests that targeting growth-factor-activated Akt in combination therapy could be a valuable therapeutic approach in treating head and neck cancer patients.
tumour microenvironment
extracellular matrix
growth factors
cell migration
Akt
head and neck cancer
1. Introduction
Cells move in response to events early in the life of all developing embryos. One of the earliest migration events is collective cell migration during gastrulation [1]. During gastrulation, cells that will become epithelial cells undergo a transition in a series of events collectively known as epithelial to mesenchymal transition (EMT) and then migrate through the primitive streak. These events are largely driven by growth factors such as platelet derived growth factor (PDGF) or fibroblast growth factor (FGF) in a phosphoinositide-3-kinase (PI3K) dependent manner [2]. PI3K, a lipid kinase, is the upstream signalling molecule of the PI3K-Akt signalling pathway which has a huge role in human development and cancer. This lipid kinase activates a membrane phospholipid, phosphatidylinositol 4,5-bisphosphate (PIP2) by phosphorylation, generating phosphatidylinositol 3,4,5-trisphosphate (PIP3). PIP3 regulates a diverse set of effector proteins including small GTPases and a group of oncogenic protein kinases called Akt or protein kinase B (PKB). Akt controls a range of cellular bioactivity including, cell growth, proliferation, survival, metabolism, and migration [3].
In general, cells that are undergoing EMT migrate by one of two main mechanisms: single cell migration or collective cell migration. During single cell migration, cells migrate as individuals having no cell–cell interactions. Two different phenotypes can be displayed during the movement of single cells. These are described as either having an amoeboid or mesenchymal phenotype. Cells with an amoeboid phenotype are generally rounded in shape with a number of different variants. The mesenchymal phenotype of cells has an elongated cell body with longer protrusions. The cells differ in terms of their contractility: amoeboid showing increased contractility (under the influence of the Rho signalling), while the mesenchymal phenotype expresses low contractility [4]. The migration of single cells through a tissue uses a multi-step process occurring through a cyclical process. The first step is protrusion from the leading edge using filopodia, lamellipodia, podosomes, or invadopodia. The second step is adhesion force generation, where the cell builds up a force strong enough to pull it through the matrix. The third step involves proteolysis in focussed areas. The fourth step being contraction of the actin–myosin cytoskeleton and finally the retraction of the rear end of the cell and its release [5]. The PI3K signalling pathway has been implicated in cell migration due to its role in controlling cytoskeletal re-arrangements and the enrichment of PtdInsP3 in the leading edge membrane of several cell types during directed cell migration [6]. It appears to be roles controlling the small GTPases, Rac and Raf, that dominate downstream of PI3K, in the control of cell autonomous migration [7]. The researchers' own data suggest a role for Akt in the migration of fibroblasts and epithelial cells [8][9]. In Chicken Embryonic Fibroblast (CEF) cells, the PI3K-Akt signalling pathway activates downstream p70S6K1 which in turn activates the Rac1 protein. Activated Rac1 is involved in actin filament remodelling, hence cell migration [10]. The PI3K-Akt pathway activates p70S6K1 in ovarian cancer, which in turn stimulates the activation of Rac1 and cdc42 and their downstream effector molecule p21 activated kinase (PAK1) [11].
Recent evidence in pancreatic ductal cancer and oesophageal squamous cell carcinoma have suggested that Akt stimulates Girdin activation which sequentially regulates actin reconstruction and cell motility [12][13].
Akt also phosphorylates Twist1 which promotes EMT by modulating its transcriptional target, TGFβ2. Increased TGFβ2 enhanced TGFβ receptor signalling, which in turn maintains hyperactive PI3K-Akt signalling [14]. Activated Akt was also found to inhibit DLC1 (Deleted in Liver Cancer 1) (GAP for RhoA), increasing levels of RhoA, facilitating the formation of focal adhesions, in turn increasing amoeboid type cell migration [15] (Figure 1).
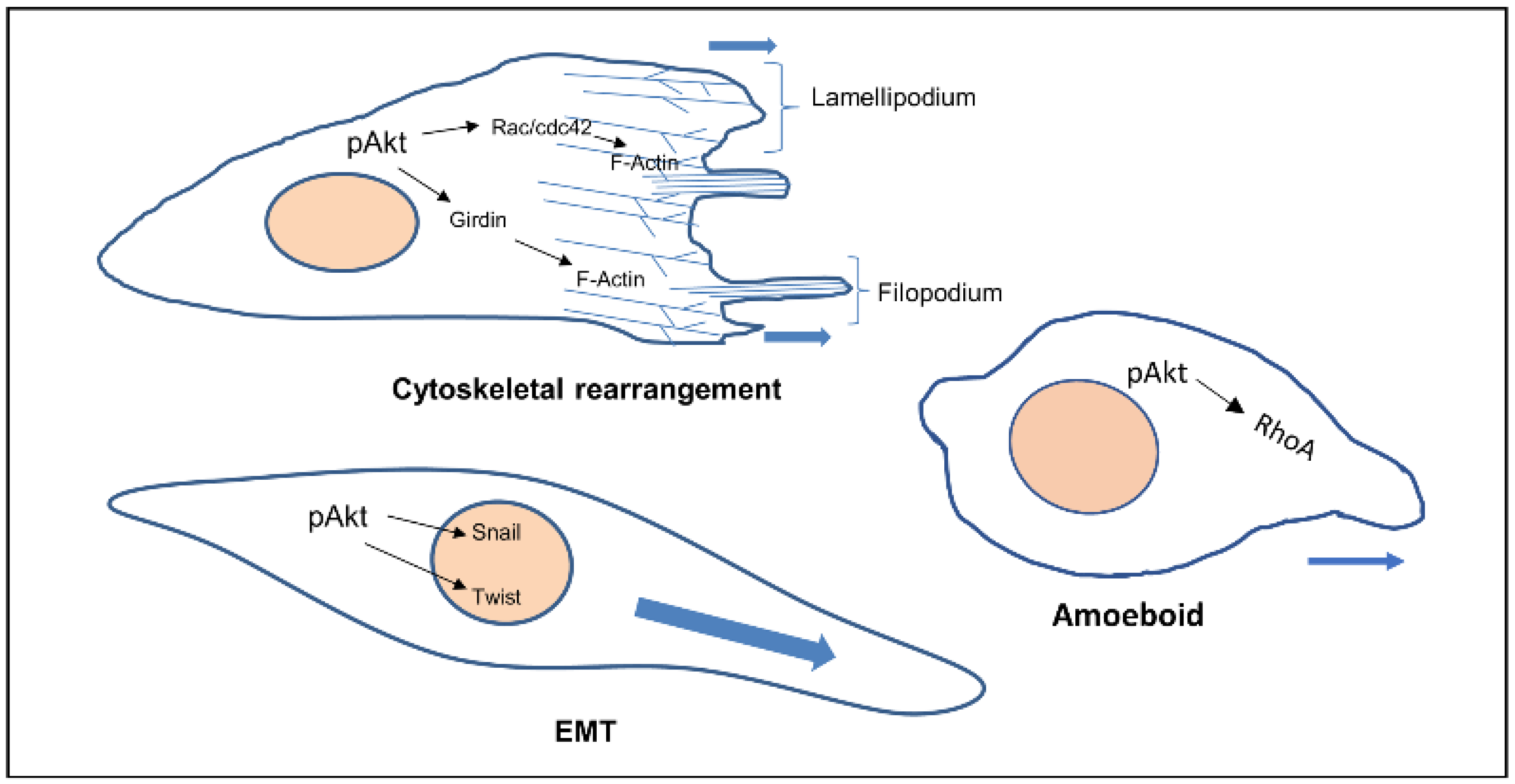
Figure 1. Cellular phenotypes during migration and the role of activated Akt. Activated Akt triggers the activity of downstream substrates (Rac, Girdin, Twist, RhoA) that dictate the various modes of cell migration.
Collective cell migration is the migration of cells as a group or sheet. The cells move in the same direction and at similar speed, it is slower but more efficient than single cell migration. There are two types of cells (leader and follower) in collective cell migration, based upon their relative position between cell clusters. The leader cell has the ability to sense the microenvironment, interact with the extracellular matrix (ECM), and is responsible for direction and speed of migration of the whole cluster. The follower cells have the ability to control their leader and are important in polarisation and chemotaxis. A successful coordination of the leaders and followers induces collective cell migration. In vivo and in vitro, leaders and follower cells can exchange their place and roles [16][17].
The ECM is the major structural component of the tissues of the body, being mainly composed of proteins, glycoproteins, glycosaminoglycans, and polysaccharides in a network. In a normal body this matrix would be laid down by connective tissue cells, such as fibroblasts. However, this tissue is also able to host tumour cells (seed and soil theory [18]) and control whether the tumour will be able to grow and metastasise. Some tissues may be able to block the ability of tumour cells moving freely through a tissue [18][19]. Therefore, the modulation of the ECM is essential for the migration of tumour cells [20]. The link between the migration of cancer cells and the spread of cancer has been established for many years and an overview produced earlier this year [21]. The original Hallmarks of Cancer included the phrase ‘tumour invasion and metastasis’ [22]. The concept of the Hallmarks of Cancer allowed new discussion around older theories, which had fallen out of mainstream thinking such as the tumour microenvironment [23]. Therefore, it is not just the cancer cells themselves that are important, it is the interactions of the tumour and the stroma that are vital. Studies into these interactions between tumour and “host” have been integral to investigating the role of growth factors (and their receptors), extracellular matrix molecules (and their receptors), and cell signalling pathways and the crosstalk between all of these factors. The tumour microenvironment (TME) is made up of a complex mixture of tumour cells and stromal-derived cells, such as cancer associated fibroblasts (CAFs), immune cells, and endothelial cells, in addition to a modified ECM. Growth factors and ECM both modulate cell signalling pathways. Human tumours are more than a mass of accumulating malignant cancer cells. Tumour cells can efficiently recruit stromal cells, immune cells, and vascular cells by secreting stimulatory growth factors, chemokines, and cytokines. These recruited cells then overexpress and release growth factors and intermediate metabolites, as well as reorganising the tissue structure to build the microenvironment. The shared communication between cancer cells and the microenvironment eventually leads to enhanced proliferation and metastasis [24] (Figure 2).
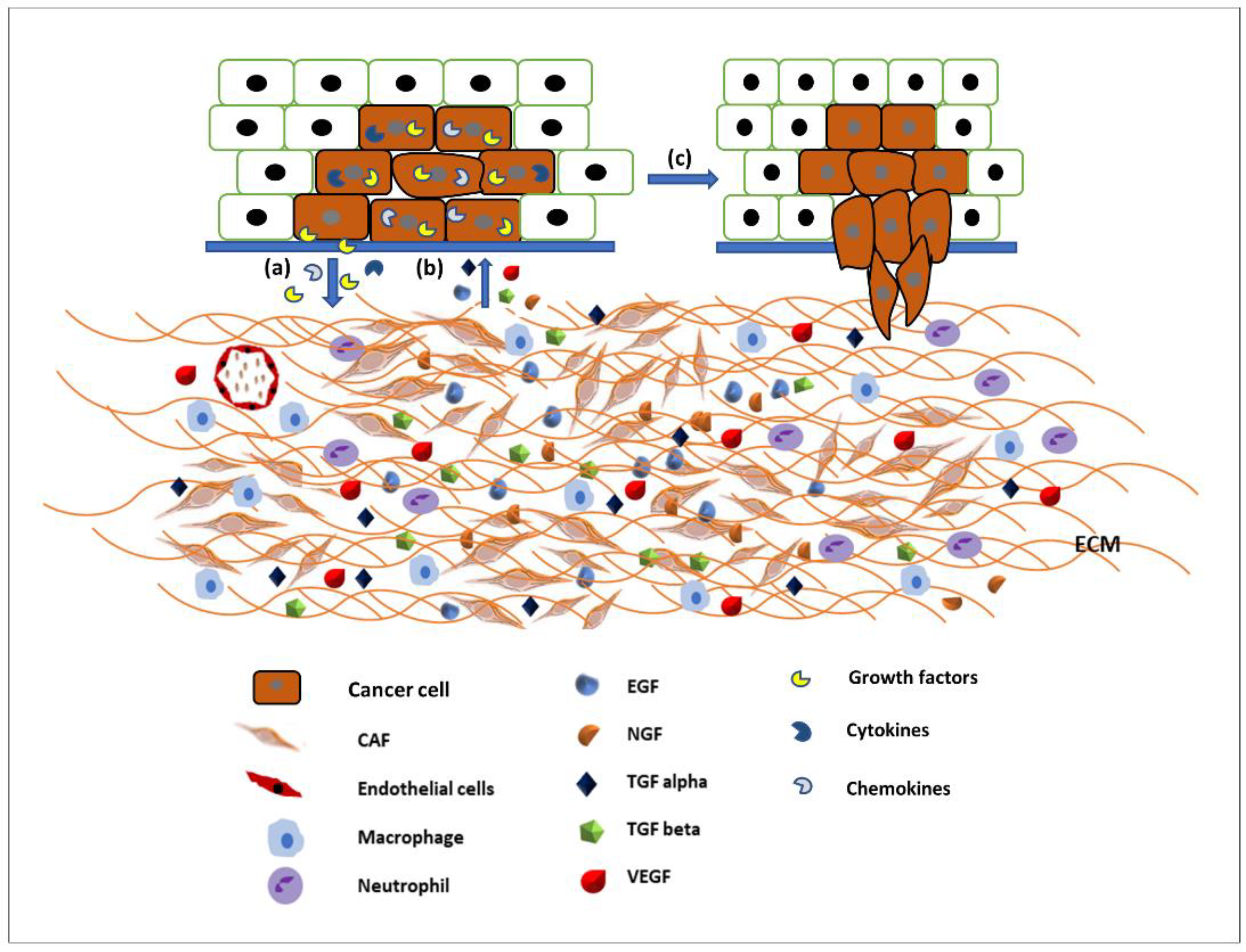
Figure 2. Communication between the tumour microenvironment and tumour cells. (a) Tumour cells release various growth factors, cytokines, and chemokines to recruit stromal cells, immune cells, and vascular cells. (b) Recruited cells then proliferation and overexpress growth factors such as EGF, TGFα and β, VEGF, NGF, etc., to initiate signalling pathways in tumour cells. (c) Activated signalling pathways then trigger various cellular activity such as tumour cell proliferation and migration and invasion.
2. Akt Signalling Pathway and Their Crosstalk in Metastasis in Head and Neck Cancer
Cetuximab, a monoclonal antibody, targeting EGFR is the only FDA-approved targeted therapy for the treatment of head and neck squamous cell carcinoma, in combination with radiation therapy or as a single agent in patients who have had prior platinum-based therapy. The response rate, as a single agent, is only 13% and the patients who respond initially eventually develop resistance [25][26]. It was hypothesised that acquired cetuximab resistance in HNSCC may result from the activation of compensatory signalling pathways following cetuximab treatment. These compensatory signalling pathways can withdraw the inhibitory effects of cetuximab through phosphorylation of key proteins, thereby promoting cell survival [27]. A protein phosphorylation profiling study showed increased phosphorylation of Akt after cetuximab treatment in acquired cetuximab-resistant cells compared to cetuximab-sensitive cells. Additionally, the study observed an additive to synergistic interaction between cetuximab and the Akt inhibitor, MK2206 in cetuximab-sensitive and acquired cetuximab-resistant HNSCC cell lines [27]. Montagut et al. showed that an EGFR mutation at S492R inhibits Cetuximab binding with the receptor but does not block EGF or TGFα binding. EGF or TGFα may therefore activate the downstream PI3K/Akt signalling pathway. Cetuximab resistance can also be mediated by the activation of the Akt signalling pathway in an alternative way, such as the overexpression of other growth factors (TGFβ, VEGF, NGF) and their associated receptors by the tumour cells and/or the tumour microenvironment [26]. Few recent clinical trials using Akt inhibitor alone to treat late stage or recurrent head and neck cancer did not show a promising outcome (Table 1).
Table 1. Recent clinical trials of Akt inhibitors in HNSCC patients. CR—complete response; PR—partial response; NPC—nasopharyngeal cancer; HNSCC—head and neck squamous cell arcinoma; ADCC—adenoid cystic carcinoma.
| Trial Identifier | Phase | Stage of Cancer | Name of the Inhibitor | Combination | Status | Result | Ref. |
|---|---|---|---|---|---|---|---|
| NCT01349933 | II | Stage IV/recurrent NPC |
MK2206 | None | Completed | CR–0% PR-4.8% Stable disease-52.4% |
[28] |
| NCT01370070 | II | Recurrent NPC |
MK2206 | None | Completed | CR–0% PR-5%, Stable disease-52% |
[29] |
| NCT01604772 | II | Recurrent/stage IV ADCC | MK2206 | None | Completed | CR/PR–0% Stable disease-81% |
[30] |
| NCT05172245 | I | Stage III-IVB HNSCC |
Ipatasertib | Cisplatin /Radiation |
Not yet recruiting | – | [31] |
It is worth noting here that activated receptor tyrosine kinases activate not only the PI3K-Akt signalling pathway, but also other pathways including MAPK and SMAD pathways. Signalling pathways are activated in a context-dependent manner and crosstalk among each other. Hence, targeted inhibition of one pathway downstream of receptors may not affect other pathways and that adds complexity to therapeutic targeting. For example, broad crosstalk exists between PI3K-Akt and Ras-Raf-MAPK pathways. Binding of ligands such as growth factors and cytokines to RTKs activates Ras. Activated Ras in turn triggers MAPK/ERK signalling pathways. Activated Ras also recruits the p110 subunit of PI3K, which in turn activates Akt signalling pathways [33][34]. Evidence also suggests that Akt can directly inhibit Raf activity by phosphorylation, hence inhibiting the MAPK pathway [35]. It has also been suggested by IP C et al. that activated Akt can trigger Rac activity by activating p70S6K [10]. Activated Rac then further activates PI3K and MAPK and acts as a bridge between PI3k-Akt and MAPK pathway crosstalk [33]. MAPKs and Akt were also found to bind and/or phosphorylate R-SMADs to control their intracellular distribution and transcriptional activity. MAPKs and Akt also phosphorylate and regulate a variety of SMAD binding partners in the nucleus, indirectly affecting the SMAD transcriptional-activation activity [36]. Other research also suggested that the PI3K-Akt signalling pathway promotes tumour metastasis by phosphorylating Twist1, via a crosstalk between Akt and TGFβ signalling [14] (Figure 3). However, crosstalk between the PI3K-Akt pathway and other receptor tyrosine-kinase-activated signalling pathways in head and neck cancer metastasis still need to be investigated.
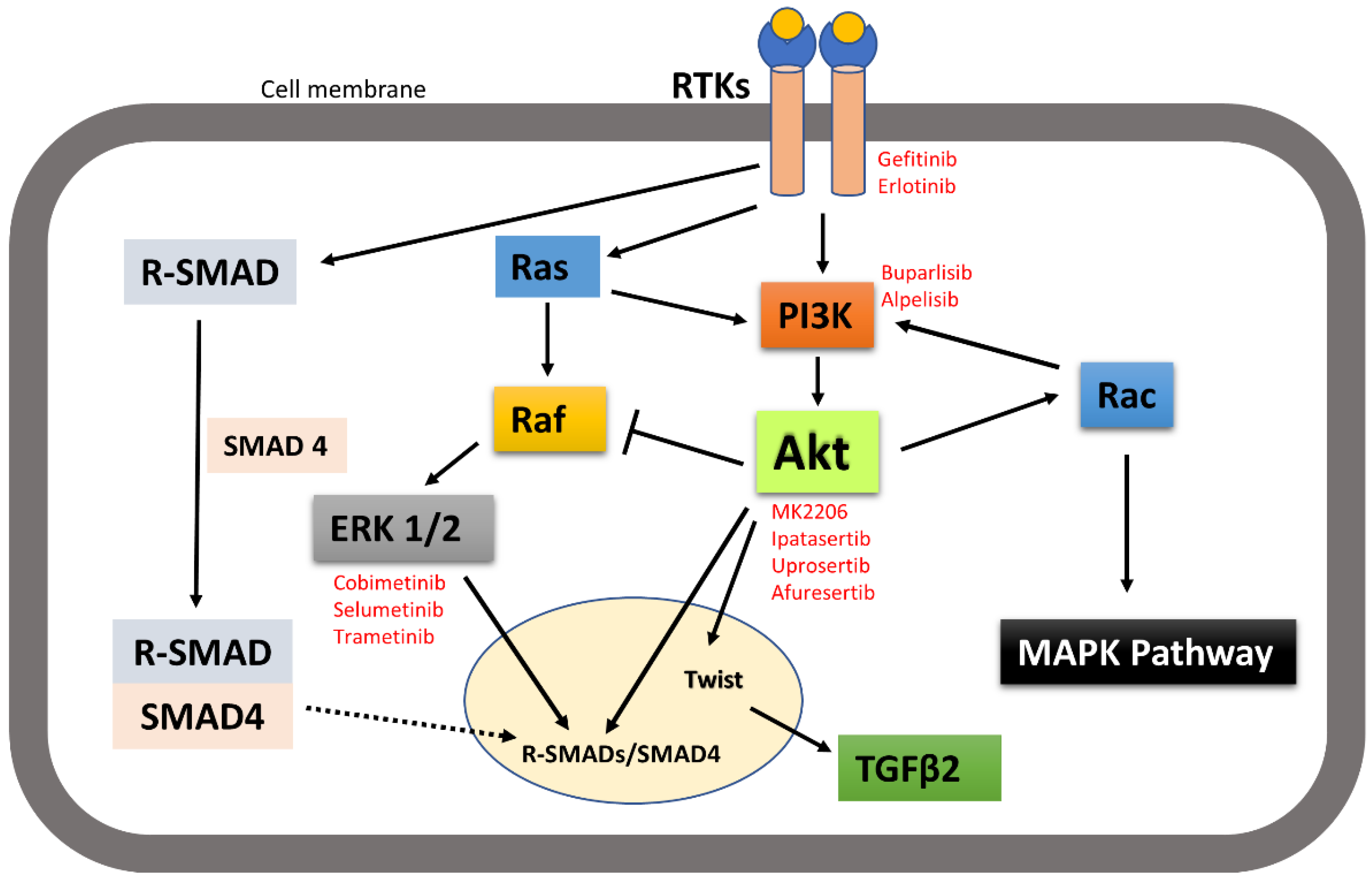
Figure 3. A simplistic graphical representation of the crosstalk between PI3K-Akt and RTK-activated other pathways. PI3K-Akt can crosstalk with MAPK and SMAD pathways to regulate their activities; thus, control cellular function. Solid arrow indicates activation, block arrow indicates inhibition, and dashed arrow indicates cellular distribution. Red texts indicate the inhibitors of the corresponding signalling molecules and receptors that are in various phases of clinical trial in treating cancer.
Published data from the researchers' group suggested that receptor tyrosine kinase inhibitors such as Gefitinib and Erlotinib inhibited the migration of head and neck cancer by inhibiting both Akt and MAPK phosphorylation [37]. Therefore, carefully designing a clinical study using a combination of an Akt inhibitor and another signalling molecule inhibitor or receptor inhibitor in the stage I–III HNSCC patient might result in an expected positive outcome.
Metastasis is a complex and multistep process in which interactions between the cellular and structural components of the TME allow cancer cells to become invasive and spread from the primary site to a distant location. Growing evidence supports the important role of the tumour microenvironment in drug resistance, as it is the main reason for the metastasis, relapse, and incurability of various cancers. Tumour-associated macrophages exert tumour growth and survival functions; CAFs reorganise the extracellular matrix creating migration-guiding tracks for cancer cells and mesenchymal cells synthesise exosomes that increase the migratory ability of the cancer cells. TME-derived exosomes are involved in the multistep process of carcinogenesis. Exosomes act as the communication channels, encouraging crosstalk between cancer and non-cancerous cells. They are associated with the increased invasiveness and drug resistance in head and neck cancer, indicative of an attractive therapeutic target [38]. Activation of the Akt pathway is one of the mechanisms involved in resistance to radiotherapy, an effective treatment modality for HNSCC [39]. Mutschelknaus et al. (2017) showed evidence that exosomes from irradiated head and neck cancer cells enhanced Akt signalling, showed increased migratory phenotype and treatment resistance compared to non-irradiated cells [40]. Thus, targeting Akt can also be an effective therapeutic strategy in exosome-mediated radiation resistant HNSCC. Akt activation is also required for T cell activation. However, evidence has shown that the sustained activation of Akt gradually drives T cells toward terminal differentiation and weakened anti-tumour activity [41]. Crompton et al. (2014) and van der Waart et al. (2014) have shown that the inhibition of Akt signalling pathway promotes generation of potent tumour-infiltrating lymphocytes (TIL) and superior tumour reactive CD8+ T cells with stem cell-like properties [42][43]. This higher proliferation capacity observed in Akt-inhibited T cells could be due to the combination of increased expression of cytokine receptors such as IL-7Rα, greater expression of co-stimulatory molecules such as CD28, and less replicative senescence characteristics [43]. Urak R et al. (2017) also revealed that Akt inhibition during ex vivo expansion did not inhibit CD19CAR (chimeric antigen receptor) T cell proliferation and effector functions [44]. Adoptive transfer of ex vivo Akt-inhibited tumour reactive CD8+ T cells and CD19CAR T cells results in increased anti-tumour effects [42][43][44].
Direct tumour–tumour cell communication, tumour–ECM interface, and tumour–stromal cell communication contributes to drug resistance. Moreover, growth factors produced in the TME provide additional signals such as Akt for cell growth, migration, and invasion and hence metastasis [45][46]. In summary, understanding the role of the tumour microenvironment in terms of activating the Akt signalling pathway and their crosstalk in metastasis in head and neck cancer, can clearly lead to the development of more effective targeted therapies, novel therapeutic combinations, or both.
References
- Weijer, C.J. Collective cell migration in development. J. Cell Sci. 2009, 122, 3215–3223.
- Geary, L.; LaBonne, C. FGF mediated MAPK and PI3K/Akt Signals make distinct contributions to pluripotency and the establishment of Neural Crest. Elife 2018, 7, e33845.
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274.
- Clark, A.G.; Vignjevic, D.M. Modes of cancer cell invasion and the role of the microenvironment. Curr. Opin. Cell Biol. 2015, 36, 13–22.
- Friedl, P.; Wolf, K. Proteolytic interstitial cell migration: A five-step process. Cancer Metastasis Rev. 2009, 28, 129–135.
- Weiner, O.D. Regulation of cell polarity during eukaryotic chemotaxis: The chemotactic compass. Curr. Opin. Cell Biol. 2002, 14, 196–202.
- Charest, P.G.; Firtel, R.A. Feedback signaling controls leading-edge formation during chemotaxis. Curr. Opin. Genet. Dev. 2006, 16, 339–347.
- Ellis, I.R.; Jones, S.J.; Lindsay, Y.; Ohe, G.; Schor, A.M.; Schor, S.L.; Leslie, N.R. Migration Stimulating Factor (MSF) promotes fibroblast migration by inhibiting AKT. Cell. Signal. 2010, 22, 1655–1659.
- Islam, M.R.; Jones, S.J.; Macluskey, M.; Ellis, I.R. Is there a pAkt between VEGF and oral cancer cell migration? Cell. Signal. 2014, 26, 1294–1302.
- Ip, C.K.M.; Cheung, A.N.Y.; Ngan, H.Y.S.; Wong, A.S.T. p70 S6 kinase in the control of actin cytoskeleton dynamics and directed migration of ovarian cancer cells. Oncogene 2011, 30, 2420–2432.
- Qian, Y.; Zhong, X.; Flynn, D.C.; Zheng, J.Z.; Qiao, M.; Wu, C.; Dedhar, S.; Shi, X.; Jiang, B.-H. ILK mediates actin filament rearrangements and cell migration and invasion through PI3K/Akt/Rac1 signaling. Oncogene 2005, 24, 3154–3165.
- Shibata, T.; Matsuo, Y.; Shamoto, T.; Hirokawa, T.; Tsuboi, K.; Takahashi, H.; Ishiguro, H.; Kimura, M.; Takeyama, H.; Inagaki, H. Girdin, a regulator of cell motility, is a potential prognostic marker for esophageal squamous cell carcinoma. Oncol. Rep. 2013, 29, 2127–2132.
- Wang, W.; Chen, H.; Gao, W.; Wang, S.; Wu, K.; Lu, C.; Luo, X.; Li, L.; Yu, C. Girdin interaction with vimentin induces EMT and promotes the growth and metastasis of pancreatic ductal adenocarcinoma. Oncol. Rep. 2020, 44, 637–649.
- Xue, G.; Restuccia, D.F.; Lan, Q.; Hynx, D.; Dirnhofer, S.; Hess, D.; Rüegg, C.; Hemmings, B.A. Akt/PKB-Mediated Phosphorylation of Twist1 Promotes Tumor Metastasis via Mediating Cross-Talk between PI3K/Akt and TGF-β Signaling Axes. Cancer Discov. 2012, 2, 248–259.
- Soriano, O.; Alcón-Pérez, M.; Vicente-Manzanares, M.; Castellano, E. The Crossroads between RAS and RHO Signaling Pathways in Cellular Transformation, Motility and Contraction. Genes 2021, 12, 819.
- Ilina, O.; Friedl, P. Mechanisms of collective cell migration at a glance. J. Cell Sci. 2009, 122, 3203–3208.
- Mayor, R.; Etienne-Manneville, S. The front and rear of collective cell migration. Nat. Rev. Mol. Cell Biol. 2016, 17, 97–109.
- Langley, R.R.; Fidler, I.J. The seed and soil hypothesis revisited—The role of tumor-stroma interactions in metastasis to different organs. Int. J. Cancer 2011, 128, 2527–2535.
- Nallanthighal, S.; Heiserman, J.P.; Cheon, D.-J. The Role of the Extracellular Matrix in Cancer Stemness. Front. Cell Dev. Biol. 2019, 7, 86.
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Primers 2020, 6, 92.
- Ellis, I.R. The Migration and Invasion of Oral Squamous Carcinoma Cells: Matrix, Growth Factor and Signalling Involvement. Cancers 2021, 13, 2633.
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Yuan, Y.; Jiang, Y.-C.; Sun, C.-K.; Chen, Q.-M. Role of the tumor microenvironment in tumor progression and the clinical applications (Review). Oncol. Rep. 2016, 35, 2499–2515.
- Khattri, A.; Sheikh, N.; Acharya, R.; Tan, Y.-H.C.; Kochanny, S.; Lingen, M.W.; Vokes, E.E.; Seiwert, T.Y. Mechanism of acquired resistance to cetuximab in head and neck cancer. J. Clin. Oncol. 2018, 36, e18061.
- Montagut, C.; Dalmases, A.; Bellosillo, B.; Crespo, M.; Pairet, S.; Iglesias, M.; Salido, M.; Gallen, M.; Marsters, S.; Tsai, S.P.; et al. Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer. Nat. Med. 2012, 18, 221–223.
- Zaryouh, H.; De Pauw, I.; Baysal, H.; Pauwels, P.; Peeters, M.; Vermorken, J.B.; Lardon, F.; Wouters, A. The Role of Akt in Acquired Cetuximab Resistant Head and Neck Squamous Cell Carcinoma: An In Vitro Study on a Novel Combination Strategy. Front. Oncol. 2021, 11, 3658.
- Institute, N.C. Akt Inhibitor MK2206 in Treating Patients with Recurrent or Metastatic Head and Neck Cancer. Available online: https://ClinicalTrials.gov/show/NCT01349933 (accessed on 4 April 2022).
- Ma, B.B.Y.; Goh, B.C.; Lim, W.T.; Hui, E.P.; Tan, E.H.; Lopes, G.d.L.; Lo, K.W.; Li, L.; Loong, H.; Foster, N.R.; et al. Multicenter phase II study of the AKT inhibitor MK-2206 in recurrent or metastatic nasopharyngeal carcinoma from patients in the mayo phase II consortium and the cancer therapeutics research group (MC1079). Investig. N. Drugs 2015, 33, 985–991.
- Ho, A.L.; Foster, N.R.; Meyers, J.P.; Vasudeva, S.D.; Katabi, N.; Antonescu, C.R.; Pfister, D.G.; Horvath, L.E.; Erlichman, C.; Schwartz, G.K. Alliance A091104: A phase II trial of MK-2206 in patients (pts) with progressive, recurrent/metastatic adenoid cystic carcinoma. J. Clin. Oncol. 2015, 33, 6039.
- Institute, N.C. Testing the Addition of Ipatasertib to Usual Chemotherapy and Radiation for Stage III-IVB Head and Neck Cancer. Available online: https://ClinicalTrials.gov/show/NCT05172245 (accessed on 4 April 2022).
- Chicago, U.O.; Institute, N.C. PI3K Inhibitor BKM120 and Cetuximab in Treating Patients with Recurrent or Metastatic Head and Neck Cancer. Available online: https://ClinicalTrials.gov/show/NCT01816984 (accessed on 19 May 2022).
- Cao, Z.; Liao, Q.; Su, M.; Huang, K.; Jin, J.; Cao, D. AKT and ERK dual inhibitors: The way forward? Cancer Lett. 2019, 459, 30–40.
- Pacold, M.E.; Suire, S.; Perisic, O.; Lara-Gonzalez, S.; Davis, C.T.; Walker, E.H.; Hawkins, P.T.; Stephens, L.; Eccleston, J.F.; Williams, R.L. Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase gamma. Cell 2000, 103, 931–943.
- Guan, K.L.; Figueroa, C.; Brtva, T.R.; Zhu, T.; Taylor, J.; Barber, T.D.; Vojtek, A.B. Negative regulation of the serine/threonine kinase B-Raf by Akt. J. Biol. Chem. 2000, 275, 27354–27359.
- Guo, X.; Wang, X.-F. Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Res. 2009, 19, 71–88.
- Thwe, A.M.; Mossey, P.; Ellis, I.R. Effect of tyrosine kinase inhibitors on cell migration and epithelial-to-mesenchymal transition in Asian head and neck cancer cell lines. J. Oral Pathol. Med. 2021, 50, 1031–1039.
- Teng, Y.; Gao, L.; Loveless, R.; Rodrigo, J.P.; Strojan, P.; Willems, S.M.; Nathan, C.-A.; Mäkitie, A.A.; Saba, N.F.; Ferlito, A. The Hidden Link of Exosomes to Head and Neck Cancer. Cancers 2021, 13, 5802.
- Bussink, J.; van der Kogel, A.J.; Kaanders, J.H.A.M. Activation of the PI3-K/AKT pathway and implications for radioresistance mechanisms in head and neck cancer. Lancet Oncol. 2008, 9, 288–296.
- Mutschelknaus, L.; Azimzadeh, O.; Heider, T.; Winkler, K.; Vetter, M.; Kell, R.; Tapio, S.; Merl-Pham, J.; Huber, S.M.; Edalat, L.; et al. Radiation alters the cargo of exosomes released from squamous head and neck cancer cells to promote migration of recipient cells. Sci. Rep. 2017, 7, 12423.
- Kim, E.H.; Suresh, M. Role of PI3K/Akt signaling in memory CD8 T cell differentiation. Front Immunol. 2013, 4, 20.
- Crompton, J.G.; Sukumar, M.; Roychoudhuri, R.; Clever, D.; Gros, A.; Eil, R.L.; Tran, E.; Hanada, K.; Yu, Z.; Palmer, D.C.; et al. Akt inhibition enhances expansion of potent tumor-specific lymphocytes with memory cell characteristics. Cancer Res. 2015, 75, 296–305.
- van der Waart, A.B.; van de Weem, N.M.P.; Maas, F.; Kramer, C.S.M.; Kester, M.G.D.; Falkenburg, J.H.F.; Schaap, N.; Jansen, J.H.; van der Voort, R.; Gattinoni, L.; et al. Inhibition of Akt signaling promotes the generation of superior tumor-reactive T cells for adoptive immunotherapy. Blood 2014, 124, 3490–3500.
- Urak, R.; Walter, M.; Lim, L.; Wong, C.W.; Budde, L.E.; Thomas, S.; Forman, S.J.; Wang, X. Ex vivo Akt inhibition promotes the generation of potent CD19CAR T cells for adoptive immunotherapy. J. Immunother. Cancer 2017, 5, 26.
- Li, Z.W.; Dalton, W.S. Tumor microenvironment and drug resistance in hematologic malignancies. Blood Rev. 2006, 20, 333–342.
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348.
More
Information
Subjects:
Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
821
Revisions:
2 times
(View History)
Update Date:
31 May 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No