Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sadegh Rajabi | -- | 3330 | 2022-05-30 11:29:24 | | | |
| 2 | Dean Liu | -3 word(s) | 3327 | 2022-05-31 03:29:15 | | | | |
| 3 | Dean Liu | Meta information modification | 3327 | 2022-05-31 03:35:52 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Rajabi, S.; Abooshahab, R.; , .; Ashrafi, M. Thyroid Cancer from the Tumor-Suppressor Genes Perspective. Encyclopedia. Available online: https://encyclopedia.pub/entry/23553 (accessed on 26 July 2026).
Rajabi S, Abooshahab R, , Ashrafi M. Thyroid Cancer from the Tumor-Suppressor Genes Perspective. Encyclopedia. Available at: https://encyclopedia.pub/entry/23553. Accessed July 26, 2026.
Rajabi, Sadegh, Raziyeh Abooshahab, , Mohammad Ashrafi. "Thyroid Cancer from the Tumor-Suppressor Genes Perspective" Encyclopedia, https://encyclopedia.pub/entry/23553 (accessed July 26, 2026).
Rajabi, S., Abooshahab, R., , ., & Ashrafi, M. (2022, May 30). Thyroid Cancer from the Tumor-Suppressor Genes Perspective. In Encyclopedia. https://encyclopedia.pub/entry/23553
Rajabi, Sadegh, et al. "Thyroid Cancer from the Tumor-Suppressor Genes Perspective." Encyclopedia. Web. 30 May, 2022.
Copy Citation
Thyroid cancer is the most frequent endocrine malignancy and accounts for approximately 1% of all diagnosed cancers. A variety of mechanisms are involved in the transformation of a normal tissue into a malignant one. Loss of tumor-suppressor gene (TSG) function is one of these mechanisms. By identifying alterations in these genes and their protein products, people can understand the thyroid cancer-related gene changes for the development of diagnostic, prognostic, and therapeutic strategies for this cancer.
thyroid cancer
tumor-suppressor gene
inactivation
diagnosis
prognosis
therapy
1. Introduction
Thyroid cancer is the most common endocrine cancer. In the United States, its incidence rate has been estimated at 12.2 cases per 100,000 individuals per year [1]. Thyroid carcinoma is the 9th most common cancer in women and the 18th cancer in both sexes, and it represents approximately 1% of all diagnosed cancers worldwide [2][3]. This cancer is classified into two histologic types: (i) follicular cell-derived cancers, such as papillary thyroid cancer (PTC; approximately 80% of all thyroid cancers), follicular thyroid cancer (FTC; 10% of all thyroid tumors), poorly differentiated thyroid cancer (4–6%), and anaplastic thyroid cancer (ATC, 2–5%) [4]; and (ii) parafollicular C cell-derived medullary thyroid carcinoma (MTC; 5–10% of all thyroid tumors) [5]. Multiple factors may play a role in their development. In breast tumors, mutations in tumor-suppressor genes (TSGs) have been identified as one of the important genetic mechanisms of breast carcinogenesis [6]. During carcinogenesis, gene alterations may affect the function of important players in normal cellular functions. These alterations may be gain-of-function mutations that particularly influence the activity of oncogenes, or loss-of-function mutations. Both may contribute to the development of the malignant phenotype [7]. TSGs play a regulatory role in cell proliferation by controlling cell-cycle progression (cell-cycle checkpoints) and consequently tissue cell proliferation. TSG deletions and other mutations may affect cancer cell proliferation, apoptosis, migration, invasion, and metastasis formation [8]. Moreover, TSGs are involved in cell differentiation regulation, genomic integrity maintenance, DNA damage repair, signaling pathways, and cell adhesion. Besides genetic alterations, other mechanisms are implicated in TSG inactivation, particularly the loss of expression due to epigenetic silencing or enhanced proteolysis of tumor-suppressor proteins [9]. These proteins are generally divided into five classes: intracellular proteins that modulate the cell cycle, hormone receptors that prevent cell proliferation, cell cycle-associated proteins that control checkpoints, apoptosis-promoting proteins, and DNA repair enzymes [10]. Studies on TSGs and their protein products are important because they may provide potential targets for thyroid cancer management. Better understanding these genes and their protein products can lead to new therapeutic applications.
2. Altered Tumor-Suppressor Genes in Thyroid Cancer
2.1. TP53
The loss of p53 function has been described in thyroid cancer. The majority of TP53 gene mutations are within exons 5–8 [11]. TP53 mutations are more frequent in ATC (50–80%) [12]. Moreover, the inactivation of p53 due to mutations or any other inhibitory mechanism may result in the progression from well-differentiated thyroid cancer to ATC [13][14]. This suggests that TP53 mutations may be a late event in thyroid cancer. In addition, some studies have shown that increased p53 protein levels are associated with thyroid cancer. Immunohistochemistry analysis revealed higher p53 protein levels in anaplastic, poorly differentiated, and well-differentiated thyroid cancer samples [13][14][15][16].
Some studies found an association between p53 and some factors involved in signaling pathways that regulate p53 expression and promote thyroid cancers. For instance, murine double minute (MDM) family members are major regulators of the p53 expression level through its ubiquitination and proteasomal degradation [17]. Prodosmo et al. showed that in PTC, the overexpression of MDM4-S, a MDM4 variant; the expression of MDM4-211, an abnormal variant; or reduced MDM4 levels all lead to p53 inactivation [18]. HECT-, UBA-, and WWE-domain-containing E3 ubiquitin protein ligase 1 (HUWE1) is an ubiquitin E3 ligase for MDM2 that can regulate p53 stabilization. In fact, HUWE1 and MDM2 are part of a network of E3 ubiquitin ligases to regulate their substrates (e.g., p53) [19]. A study of some thyroid cancer cell lines to assess HUWE1 function demonstrated that HUWE1 downregulation leads to MDM2 overexpression and decreased p53 protein stability, suggesting that it may act as a TSG [20]. In addition, other factors play a role in p53 regulation, such as wild-type p53-induced phosphatase 1 (WIP1), the overexpression of which can inhibit p53 in PTCs [21]. In an animal study, Zou et al. found that TP53 downregulation leads to higher levels of thyroid-stimulating hormone (TSH) and the subsequent upregulation of the PI3K/AKT pathway. This was associated with PTC-to-ATC transformation and ATC progression [22]. Altogether, the data mentioned above suggest a dual function of TP53 as an oncogene and TSG.
2.2. PTEN
PTEN is a TSG associated with the negative regulation of signaling pathways, such as the PI3K signaling cascade [23]. The loss of PTEN activity in Cowden syndrome increases the risk of some cancers, including thyroid cancer [24]. Patients with this syndrome are at a higher risk of developing FTC due to pathogenic mutations in the PTEN gene [25]. PTEN is considered a predictive marker in patients with thyroid cancer and Cowden-like syndrome. For instance, Ngeow et al. showed that very low serum levels of PTEN in these patients could predict the presence of a germline PTEN mutation [26]. PTEN inactivation might be the result of different mechanisms, such as point mutations, deletions, promoter hypermethylation, and post-translational modifications [27][28][29][30]. PTEN somatic deletions and LOH have also been described in many tumor types, especially in thyroid cancer subtypes [31].
PTEN hamartoma tumor syndrome (PHTS) is a complex disease caused by germline PTEN gene mutations. These patients usually develop various benign and malignant tumors in different tissues, such as breast, thyroid, intestine, and skin [32]. The lifetime risk of thyroid cancer in patients with PHTS who have a PTEN mutation has been estimated at 35.2% [33].
Moreover, Nagy et al. found that 3–10% of patients with PTEN mutations have differentiated thyroid cancer (DTC) [34]. Specifically, in a sample of patients with DTC, 4.8% of patients with FTC harbored germline PTEN mutations, but none of the patients with PTC did [34]. Other studies also reported the presence of PTEN mutations in FTC tissue samples [35][36]. However, sporadic thyroid cancers do not harbor somatic PTEN mutations [37].
PTEN is also regulated through various epigenetic mechanisms. For example, aberrant PTEN methylation impairs PTEN function, leading to enhanced PI3K/AKT signaling, and thyroid cancer growth, progression, and metastasis formation [38]. Methylation-specific polymerase-chain reaction analysis of 59 thyroid cancer samples showed that the PTEN promoter was hypermethylated in 45.7% of PTCs, 85% of FTCs, and 83% of follicular adenomas [39]. Pringle et al. developed mouse models in which two TSGs, protein kinase CAMP-dependent type 1 regulatory subunit alpha (Prkar1a) and Pten, were concomitantly knocked out in the thyroid. They found that this double knockout led to FTC development with metastatic spread, and enhanced function of protein kinase A (PKA) and mammalian target of rapamycin (mTOR) signaling [40]. Some studies suggested that the loss of PTEN expression is associated with the low expression of p27, a cell-cycle inhibitor, in FTC and ATC specimens [41][42].
Ubiquitination also might be implicated in PTEN inhibition. Yu et al. found that two succinate dehydrogenase-D variants, G12S and H50R, induce PTEN mono-ubiquitination, leading to its translocation into the nucleus where PTEN causes AKT upregulation and promotes tumorigenesis via FOXO3a phosphorylation and autophagy downregulation [43]. Frisk et al. observed no relationship between PTEN expression, LOH, and mutation status in the ATC samples under study. They suggested that PTEN inactivation may be due to a number of epigenetic and/or structural silencing mechanisms rather than to classical biallelic inactivation [44].
2.3. APC
Germline mutations in the APC gene lead to familial adenomatous polyposis (FAP) syndrome [45]. Most APC mutations are nonsense and frameshift mutations [46]. In 1949, Crail described for the first time the development of thyroid cancer in patients with FAP [47]. Based on the Leeds Castle Polyposis Group database, the incidence of thyroid cancer in patients with FAP is 1.2% [48]. The mean age at diagnosis of thyroid cancer in these patients is between 25 and 28 years [48][49]. PTC frequency in patients with FAP is higher in women than in men (10–17:1) [50][51].
APC mutations are rarely detected in sporadic thyroid cancers and may play a role in thyroid cancer development [52]. Moreover, APC mutations are associated with RET/PTC1 gene rearrangements in patients with FAP and thyroid cancer [53]. APC gene mutation analysis in a 25-year-old woman with FAP and previous total colectomy and thyroidectomy revealed the presence of a germline mutation in exon 13 [54]. Exons 1–15 of the APC gene and exon 3 of the beta-catenin gene were analyzed in genomic DNA extracted from peripheral blood samples and 12 cribriform-morular variants of papillary thyroid carcinoma nodules. Besides the germ-line mutation, six somatic mutations between codons 308 and 935 of the APC gene were found, but none were found in the beta-catenin gene [54]. Cetta et al. reported that 13/15 patients with FAP and thyroid cancer carried APC mutations between codons 778 and 1309 [55]. Truta et al. analyzed APC in 14 patients with FAP and PTC and identified germline mutations (located before codon 1286 and outside the APC mutation cluster region) in 12 patients [56]. Another study showed that the risk of developing thyroid cancer is higher in individuals with APC mutations at the 5’ end, near codon 528, and that this risk further increases in subjects harboring a mutation at codon 1061 [57]. Han et al. carried out an APC mutation screening in Korean patients with FAP and identified nine truncating mutations, one missense mutation, seven polymorphisms, and three intronic variants [58]. In a European cooperative study, the analysis of APC mutations in 15 women with FAP and PTC uncovered APC germline mutations in 13 of them at codons 140, 593, 778, 976, 993, 1061 (n = 5), 1105 (n = 1), and 1309 (n = 2) [49].
Some data indicate that epigenetic mechanisms also are implicated in the regulation of APC expression. Mir-155 can target APC by direct binding to its 3’-untranslated region, and this may decrease APC mRNA and protein levels [59]. Zhang et al. found that upregulation of mir-155 results in APC downregulation and the activation of WNT/β-catenin signaling, which in turn promotes PTC cell growth, viability, and colony formation. This suggests that mir-155 may play an oncogenic role in these tumors [59]. APC protein has a binding site for β-catenin that consists of seven 20-amino acid repeats (20-AARs) [60]. Kumamoto et al. carried out a retrospective study to find an association between the number of 20-A ARs and thyroid cancer development [61]. They observed that in three patients with FAP and PTC, one patient had only two 20-AARs in the germline APC mutation and none in the somatic APC mutation, and the other two did not have any remaining 20-AAR. Moreover, in 13/16 patients with FAP and thyroid cancer (81.3%), the remaining number of 20-AARs was zero. Therefore, they suggested that in patients with FAP and thyroid cancer, the APC/β-catenin signaling pathway may play an important role in the pathogenesis of this cancer [61].
2.4. RASAL1
RAS protein activator like 1 (RASAL1) is the negative modulator of the RAS signaling pathway that has been identified as a key TSG in thyroid carcinoma [62]. The RAS-coupled mitogen-activated protein kinase (MAPK) and PI3K pathways play pivotal roles in thyroid cancer development. Therefore, abnormal RASAL1 gene expression may affect these pathways and thyroid cancer development [63]. In an in vitro and in vivo study, Lui et al. investigated alterations of genes encoding negative modulators of the RAS-coupled MAPK and PI3K pathways in thyroid cancer. They discovered RASAL1 gene-disabling mutations and promoter hypermethylation in thyroid cancer samples, predominantly FTC and ATC, suggesting the RASAL1 is a key TSG in thyroid cancer. They found one nonsense and six missense mutations that were located at highly conserved sites within the RAS GTPase activating domain of RASAL1 [63].
Ngeow et al. investigated the presence of germline RASAL1 mutations and PTEN mutation status in patients with Cowden syndrome and thyroid cancer. Among the included 155 patients, they identified deleterious RASAL1 germline mutations in two patients with wild-type PTEN who had FTC. They also detected detrimental germline RASAL1 mutations in patients with Cowden syndrome and follicular-variant PTC, unlike many other patients with Cowden syndrome [64]. Charalampos et al. described a patient with MTC, mesothelioma, and meningioma who harbored APC and RASAL1 mutations based on whole exome sequencing (WES) data. This indicates a possible TSG role for both APC and RASAL1 in thyroid cancer development [65]. Moreover, WES data indicate the presence of APC and RASAL1 gene alterations in various thyroid cancer subtypes. For example, WES and Sanger sequencing of a MTC sample from a 57-year-old woman with sporadic MTC showed two germline APC and RASAL1 variants [66]. Moreover, the targeted exome sequencing of DNA from 11 formalin-fixed, paraffin-embedded ATC tissue samples uncovered two specimens (18%) with a RASAL1 mutation that was significantly associated with shorter survival [67].
2.5. TP63 and TP73
Several studies found a possible role for p63, a p53 homolog, in thyroid cancer development [68][69]. TP63 is expressed in epithelia of ectodermal origin. This gene has six different isoforms with a transactivating effect or dominant negative activity on p53 target genes [68]. Bonzanini et al. reported p63 immunostaining in PTC samples but not in controls [70]. In another study, TAp63α, an isoform of the TP63 gene, was detected in thyroid cancer samples. The authors suggested that it may act as an oncogene by promoting thyroid cancer progression via the disruption of p53 tumor-suppressor activity [71]. However, TAp63β and TAp63γ (two other p63 isoforms) have tumor-suppressor activity in PTC and FTC cells [71]. These data show that TP63, like TP53, may play a dual role in thyroid cancer: oncogene and TSG. P73 is another member of the p53 family that plays a controversial role in thyroid cancer. Ferru et al. reported that the expression of TAp73 and DNp73, two p73 isoforms, is reduced in follicular adenomas, FTCs, and PTCs. The TAp73 variant has pro-apoptotic, and DNp73 has anti-apoptotic, properties [72].
Some immunohistochemical studies revealed that p73 and DNp73 are expressed in human thyroid cancer specimens. These results were also confirmed by RT-PCR analysis showing that DNp73a is expressed in malignant thyroid tissues but not in normal tissues [73][74]. Periostin is a mesenchyme-specific protein that, when overexpressed, is associated with aggressive forms of thyroid cancer [75]. Puppin et al. suggested that DNp73α could induce periostin gene expression in papillary, follicular, and undifferentiated thyroid cancer cells [76]. Vella et al. showed that DNp73α overexpression in thyroid cancer cells leads to decreased PTEN expression [77]. Conversely, Malaguarnera et al. suggested that in thyroid cancer cells, TAp73α promotes p53 protein expression by inhibiting MDM2-mediated p53 degradation, proposing a thyroid-specific dual function for this TSG [71].
2.6. RB
Dysregulated RB expression in thyroid cancer has been demonstrated in several previous studies [78][79][80]. RB mutations and other inactivating mechanisms may play a role in thyroid cancer pathogenesis, especially MTC [78]. The cyclin-dependent-kinase inhibitor (CDKI)-RB1 pathway plays a pivotal role in the control of cell-cycle checkpoints. Loss of CDKIs leads to increased RB phosphorylation and consequently uncontrolled cell-cycle progression. Some studies showed the impairment of this pathway, which can promote MTC tumorigenesis [81][82]. In a conditional mouse model, Pozo et al. found that CDK5 overactivation in C-cells promotes sporadic forms of MTC through Rb downregulation [83]. Gilbert et al. observed that the inactivation of RB1 regulatory pathways in parafollicular C-cells leads to a high rate of MTC in mouse models [82]. Similarly, Rb1 deletion may induce MTC in mice [84]. By immunohistochemistry, Valenciaga et al. demonstrated that RB expression reduction is associated with aggressive MTC [85]. More interestingly, the loss of RB expression in PTC samples has been correlated with aggressive early-metastasizing forms of this thyroid cancer [86].
2.7. PRKAR1A
The PRKAR1A gene encodes the regulatory subunit 1 alpha of PKA and is involved in the PKA signaling pathway. The binding of ligands to their receptors at the cell membrane could cause the activation of G-proteins that then induce adenylyl cyclase enzyme activity and convert AMP to cyclic AMP (cAMP). Upon cAMP binding to regulatory subunits, the PKA enzyme is activated and phosphorylates serine and threonine residues on substrate proteins. The cAMP response element (CRE)-binding protein (CREBP) is a transcription factor that is phosphorylated by PKA and translocated into the nucleus, where it binds to CREs and upregulates the expression of different genes. Through this kinase activity, PKA could regulate different cellular processes, including growth, division, and differentiation. TSH is one of the ligands that could activate PKA via its receptor. Elevated TSH could contribute to the development of thyroid cancer [87]. On the other hand, the phosphatase PTEN could dephosphorylate the CREBP transcription factor in the nucleus [88]. Zhang and colleagues showed that the inhibition of phosphodiesterase 4, which is responsible for cAMP degradation, could lead to TP53 upregulation, followed by cancer cell proliferation inhibition and apoptosis induction [89]. PKA induction (via PRKAR1A downregulation), through adrenaline receptor activation, and CHEK2 downregulation by the glucocorticoid receptor, could inhibit the DNA damage response, leading to the dysregulation of the DNA repair system; apoptosis; and cell-cycle checkpoint deregulation and carcinogenesis [90].
The Carney complex is a familial neoplasia syndrome associated with thyroid tumors. PRKAR1A is often mutated in this syndrome [91]. Moreover, LOH for the PRKAR1A locus has been reported in undifferentiated thyroid cancers (ATC) [92]. Pringle et al. showed that Prkar1a knockout in the thyroid can lead to hyperthyroidism and FTC development in mice [93]. They also developed mice in which both PRKAR1A and PTEN are knocked out and found that these animals develop FTC with a metastatic phenotype [40].
2.8. CHEK2
CHEK2 is one of the most important genes involved in cell-cycle control by encoding the human analog of the yeast checkpoint kinases Cds1 and Rad53 [94]. CHEK2 acts as a protective regulator of cell division in response to DNA damage by preventing cell entry into mitosis. This suggests that CHEK2 might be a TSG. Abnormalities in cell proliferation and apoptotic evasion due to CHEK2 gene mutations have been observed in various cancers [95][96][97]. In a study on the association of CHEK2 mutations with different cancer types in Poland, the most significant correlation was observed between thyroid cancer (mainly PTC) and CHEK2 protein-truncating mutations [98]. Ten years later, Wójcicka et al. investigated deleterious polymorphisms in ATM, CHEK2, and BRCA1 in 1,781 patients with PTC and 2081 healthy controls using the Sequenom technology. They found that the CHEK2 rs17879961 variant is associated with increased PTC risk [99]. However, Fayaz et al. analyzed the two most common CHEK2 gene mutations in 100 DTC samples and found that they are not associated with higher DTC risk in the Iranian population [100]. This suggests that these two mutations are not detrimental and do not inactivate CHEK2. Kaczmarek-Ryś et al. used a genotyping technique to identify specific CHEK2 alterations in 602 patients with DTC and 829 controls [101]. They found that the CHEK2 c.470C variant increases the risk of DTC in the Polish population. Another genetic study to estimate the somatic alteration profile in FTC samples using next-generation sequencing identified CHEK2 gene alterations in FTC samples [102].
The different TSGs altered and implicated in the pathogenesis of thyroid cancer are shown in Table 1. The molecular pathways that are regulated by these TSGs are depicted in Figure 1.
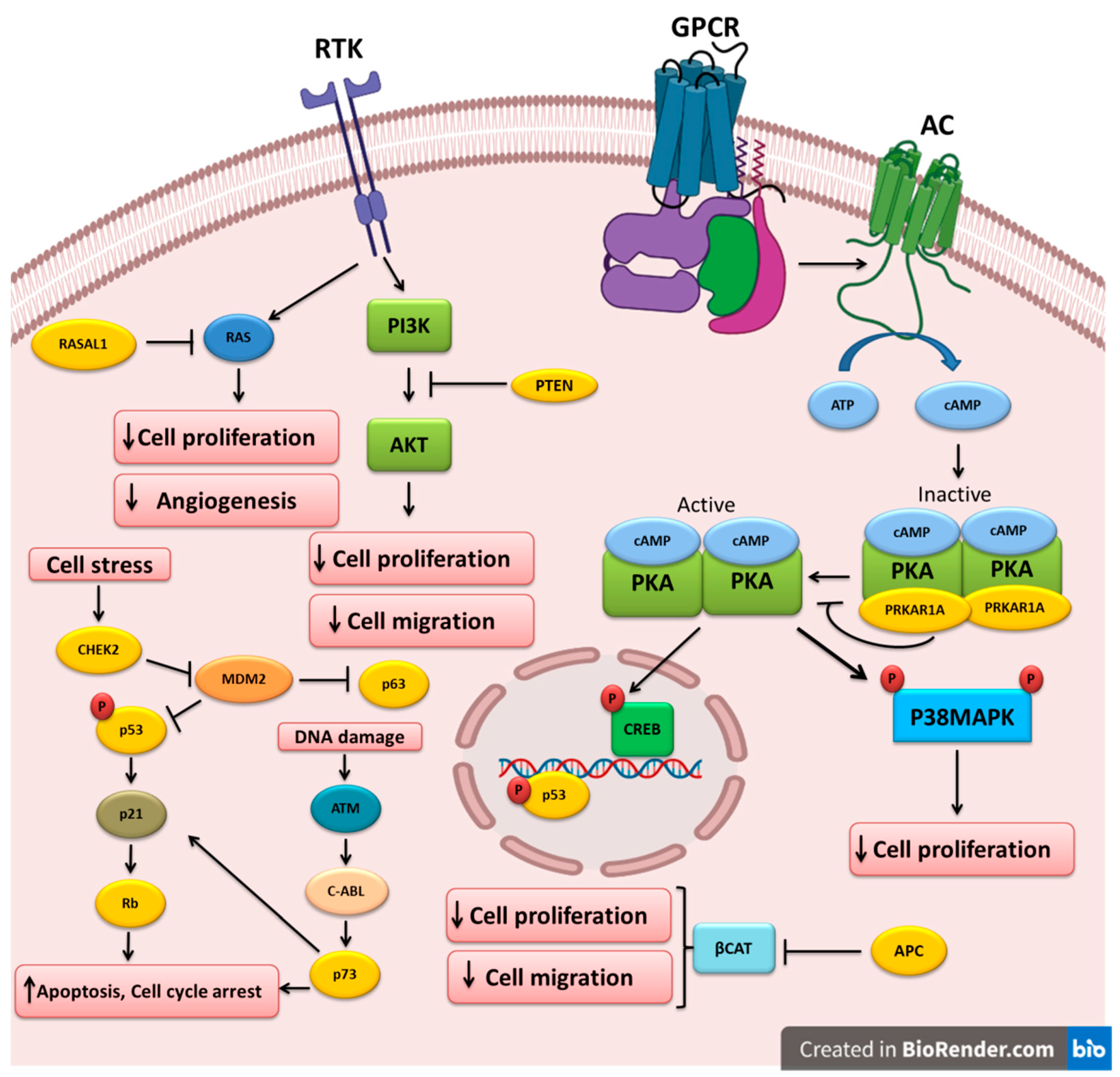
Figure 1. Key tumor-suppressor genes and their related pathways in thyroid cancer. Growth factors bind to their receptor tyrosine kinases (RTK) and activate the PI3K and RAS pathways that could be inhibited by PTEN and RASAL1, respectively. Cell stress triggers CHEK2, which in turn downregulates MDM2, leading to the upregulation of the p53 and p63 tumor-suppressors. P53 can also increase RB expression. DNA damage could lead to p73 upregulation and apoptosis induction. APC inhibits beta-catenin, leading to the inhibition of cell proliferation and migration. Ligand binding to G-protein-coupled receptors (GPCR) induces G-protein and adenylyl cyclase (AC), which increases cAMP and PKA activation. PRKAR1A mutations could cause PKA hyperactivation and cell proliferation induction.
Table 1. Altered tumor-suppressor genes involved in thyroid cancer pathogenesis.
| Tumor Suppressor Gene | Normal Function of Protein Product | Type of Alterations | Affected Thyroid Tumors | Mutation Frequency |
|---|---|---|---|---|
| TP53 | Cell-cycle regulation | Point mutations, negative regulation by MDM family members, and ubiquitination. Dual function: oncogene and TSG | ATC (50–80%), PTC | 40% in PTC, 60% in ATC |
| PTEN | Cell division regulation | Point mutations, deletion, promoter hypermethylation, LOH, ubiquitination, and post-translational modifications | FTC, DTC, ATC, and PTC | 65–85% |
| APC | The regulation of cell division, adhesion, and migration | Nonsense and missense mutations, frameshift mutations, polymorphisms, and epigenetic regulation | ATC, MTC, PTC, FTC, and CMVPTC | 87% in FAP-associated PTC |
| RASAL1 | The stimulation of the GTPase activity of RAS | Missense and nonsense mutations, promoter hypermethylation | PTC, ATC, FTC, and MTC | 17% in ATCs, 5% in FTCs, and 3% in PTCs |
| TP63 | The regulation of cell proliferation and differentiation, the transactivating effect, or dominant negative activity on p53 target genes | Downregulation, dual function: oncogene and TSG | PTC and FTC | |
| TP73 | Involved in cellular responses to stress and development | Downregulation and upregulation, dual function: oncogene and TSG | Follicular adenoma, FTC, and PTC | |
| RB | The control of DNA replication and cell division during cell damage | Mutations, deletions, downregulation, enhanced phosphorylation, and the loss of expression | MTC, PTC | 1.8% in MTC |
| PRKAR1A | Promoting cell growth and division | Downregulation, LOH | FTC, ATC | LOH of the PRKAR1A(CA)n locus in 37.5% of cases |
| CHEK2 | Cell-cycle control | Mutations, polymorphisms | PTC, FTC, and DTC | 15.2% |
PTC, papillary thyroid cancer; FTC, follicular thyroid cancer; ATC, anaplastic thyroid cancer; MTC, medullary thyroid carcinoma; DTC, differentiated thyroid carcinoma; CMVPTC, cribriform-morular variant of papillary thyroid carcinoma; and LOH, loss of heterozygosity.
References
- Rajabi, S.; Dehghan, M.H.; Dastmalchi, R.; Jalali Mashayekhi, F.; Salami, S.; Hedayati, M. The roles and role-players in thyroid cancer angiogenesis. Endocr. J. 2019, 66, 277–293.
- Vianna, D.M.; Curioni, O.A.; Franca, L.J.; Paiva, D.L.; Pompeu, B.F.; Dedivitis, R.A.; Rapoport, A. The histological rarity of thyroid cancer. Braz. J. Otorhinolaryngol. 2012, 78, 48–51.
- Mendelsohn, J.; Howley, P.M.; Israel, M.A.; Gray, J.W.; Thompson, C. The Molecular Basis of Cancer, 4th ed.; Elsevier Inc.: New York, NY, USA, 2015; pp. 835–863.
- Lee, J.; Hwang, J.A.; Lee, E.K. Recent progress of genome study for anaplastic thyroid cancer. Genom. Inform. 2013, 11, 68–75.
- Rajabi, S.; Hedayati, M. Medullary Thyroid Cancer: Clinical Characteristics and New Insights into Therapeutic Strategies Targeting Tyrosine Kinases. Mol. Diagn. Ther. 2017, 21, 607–620.
- Buchholz, T.A.; Weil, M.M.; Story, M.D.; Strom, E.A.; Brock, W.A.; McNeese, M.D. Tumor suppressor genes and breast cancer. Radiat. Oncol. Investig. 1999, 7, 55–65.
- Osborne, C.; Wilson, P.; Tripathy, D. Oncogenes and tumor suppressor genes in breast cancer: Potential diagnostic and therapeutic applications. Oncologist 2004, 9, 361–377.
- Maziveyi, M.; Alahari, S.K. Breast Cancer Tumor Suppressors: A Special Emphasis on Novel Protein Nischarin. Cancer Res. 2015, 75, 4252–4259.
- Oliveira, A.M.; Ross, J.S.; Fletcher, J.A. Tumor suppressor genes in breast cancer: The gatekeepers and the caretakers. Am. J. Clin. Pathol. 2005, 124, S16–S28.
- Lodish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Dardell, J. Proto-Oncogenes and Tumor-Suppressor Genes. In Molecular Cell Biology, 4th ed.; Freeman & Company: New York, NY, USA, 2000; pp. 845–867.
- Farid, N.R. P53 mutations in thyroid carcinoma: Tidings from an old foe. J. Endocrinol. Investig. 2001, 24, 536–545.
- Dralle, H.; Machens, A.; Basa, J.; Fatourechi, V.; Franceschi, S.; Hay, I.D.; Nikiforov, Y.E.; Pacini, F.; Pasieka, J.L.; Sherman, S.I. Follicular cell-derived thyroid cancer. Nat. Rev. Dis. Primers 2015, 1, 15077.
- Pollina, L.; Pacini, F.; Fontanini, G.; Vignati, S.; Bevilacqua, G.; Basolo, F. bcl-2, p53 and proliferating cell nuclear antigen expression is related to the degree of differentiation in thyroid carcinomas. Br. J. Cancer 1996, 73, 139–143.
- Donghi, R.; Longoni, A.; Pilotti, S.; Michieli, P.; Della Porta, G.; Pierotti, M.A. Gene p53 mutations are restricted to poorly differentiated and undifferentiated carcinomas of the thyroid gland. J. Clin. Investig. 1993, 91, 1753–1760.
- Park, K.Y.; Koh, J.M.; Kim, Y.I.; Park, H.J.; Gong, G.; Hong, S.J.; Ahn, I.M. Prevalences of Gs alpha, ras, p53 mutations and ret/PTC rearrangement in differentiated thyroid tumours in a Korean population. Clin. Endocrinol. 1998, 49, 317–323.
- Soares, P.; Cameselle-Teijeiro, J.; Sobrinho-Simoes, M. Immunohistochemical detection of p53 in differentiated, poorly differentiated and undifferentiated carcinomas of the thyroid. Histopathology 1994, 24, 205–210.
- Chen, D.; Li, M.; Luo, J.; Gu, W. Direct interactions between HIF-1 alpha and Mdm2 modulate p53 function. J. Biol. Chem. 2003, 278, 13595–13598.
- Prodosmo, A.; Giglio, S.; Moretti, S.; Mancini, F.; Barbi, F.; Avenia, N.; Di Conza, G.; Schunemann, H.J.; Pistola, L.; Ludovini, V.; et al. Analysis of human MDM4 variants in papillary thyroid carcinomas reveals new potential markers of cancer properties. J. Mol. Med. 2008, 86, 585–596.
- Kurokawa, M.; Kim, J.; Geradts, J.; Matsuura, K.; Liu, L.; Ran, X.; Xia, W.; Ribar, T.J.; Henao, R.; Dewhirst, M.W.; et al. A network of substrates of the E3 ubiquitin ligases MDM2 and HUWE1 control apoptosis independently of p53. Sci. Signal. 2013, 6, ra32.
- Ma, W.; Zhao, P.; Zang, L.; Zhang, K.; Liao, H.; Hu, Z. Tumour suppressive function of HUWE1 in thyroid cancer. J. Biosci. 2016, 41, 395–405.
- Yang, D.; Zhang, H.; Hu, X.; Xin, S.; Duan, Z. Abnormality of pl6/p38MAPK/p53/Wipl pathway in papillary thyroid cancer. Gland Surg. 2012, 1, 33–38.
- Zou, M.; Baitei, E.Y.; Al-Rijjal, R.A.; Parhar, R.S.; Al-Mohanna, F.A.; Kimura, S.; Pritchard, C.; Binessa, H.A.; Alzahrani, A.S.; Al-Khalaf, H.H.; et al. TSH overcomes Braf(V600E)-induced senescence to promote tumor progression via downregulation of p53 expression in papillary thyroid cancer. Oncogene 2016, 35, 1909–1918.
- Xing, M. Genetic alterations in the phosphatidylinositol-3 kinase/Akt pathway in thyroid cancer. Thyroid 2010, 20, 697–706.
- Eng, C. The role of PTEN, a phosphatase gene, in inherited and sporadic nonmedullary thyroid tumors. Recent Prog. Horm. Res. 1999, 54, 441–452.
- Ngeow, J.; Mester, J.; Rybicki, L.A.; Ni, Y.; Milas, M.; Eng, C. Incidence and clinical characteristics of thyroid cancer in prospective series of individuals with Cowden and Cowden-like syndrome characterized by germline PTEN, SDH, or KLLN alterations. J. Clin. Endocrinol. Metab. 2011, 96, E2063–E2071.
- Ngeow, J.; He, X.; Mester, J.L.; Lei, J.; Romigh, T.; Orloff, M.S.; Milas, M.; Eng, C. Utility of PTEN protein dosage in predicting for underlying germline PTEN mutations among patients presenting with thyroid cancer and Cowden-like phenotypes. J. Clin. Endocrinol. Metab. 2012, 97, E2320–E2327.
- Goschzik, T.; Gessi, M.; Denkhaus, D.; Pietsch, T. PTEN mutations and activation of the PI3K/Akt/mTOR signaling pathway in papillary tumors of the pineal region. J. Neuropathol. Exp. Neurol. 2014, 73, 747–751.
- Lotan, T.L.; Carvalho, F.L.; Peskoe, S.B.; Hicks, J.L.; Good, J.; Fedor, H.; Humphreys, E.; Han, M.; Platz, E.A.; Squire, J.A.; et al. PTEN loss is associated with upgrading of prostate cancer from biopsy to radical prostatectomy. Mod. Pathol. 2015, 28, 128–137.
- Piras, G.; Monne, M.; Palmas, A.D.; Calvisi, A.; Asproni, R.; Vacca, F.; Pilo, L.; Gabbas, A.; Latte, G. Methylation analysis of the phosphates and tensin homologue on chromosome 10 gene (PTEN) in multiple myeloma. Clin. Epigenet. 2014, 6, 16.
- Yang, Z.; Yuan, X.G.; Chen, J.; Luo, S.W.; Luo, Z.J.; Lu, N.H. Reduced expression of PTEN and increased PTEN phosphorylation at residue Ser380 in gastric cancer tissues: A novel mechanism of PTEN inactivation. Clin. Res. Hepatol. Gastroenterol. 2013, 37, 72–79.
- Dahia, P.L.; Marsh, D.J.; Zheng, Z.; Zedenius, J.; Komminoth, P.; Frisk, T.; Wallin, G.; Parsons, R.; Longy, M.; Larsson, C.; et al. Somatic deletions and mutations in the Cowden disease gene, PTEN, in sporadic thyroid tumors. Cancer Res. 1997, 57, 4710–4713.
- Smith, J.R.; Marqusee, E.; Webb, S.; Nose, V.; Fishman, S.J.; Shamberger, R.C.; Frates, M.C.; Huang, S.A. Thyroid nodules and cancer in children with PTEN hamartoma tumor syndrome. J. Clin. Endocrinol. Metab. 2011, 96, 34–37.
- Tan, M.H.; Mester, J.L.; Ngeow, J.; Rybicki, L.A.; Orloff, M.S.; Eng, C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin. Cancer Res. 2012, 18, 400–407.
- Nagy, R.; Ganapathi, S.; Comeras, I.; Peterson, C.; Orloff, M.; Porter, K.; Eng, C.; Ringel, M.D.; Kloos, R.T. Frequency of germline PTEN mutations in differentiated thyroid cancer. Thyroid 2011, 21, 505–510.
- Gimm, O.; Perren, A.; Weng, L.P.; Marsh, D.J.; Yeh, J.J.; Ziebold, U.; Gil, E.; Hinze, R.; Delbridge, L.; Lees, J.A.; et al. Differential nuclear and cytoplasmic expression of PTEN in normal thyroid tissue, and benign and malignant epithelial thyroid tumors. Am. J. Pathol. 2000, 156, 1693–1700.
- Halachmi, N.; Halachmi, S.; Evron, E.; Cairns, P.; Okami, K.; Saji, M.; Westra, W.H.; Zeiger, M.A.; Jen, J.; Sidransky, D. Somatic mutations of the PTEN tumor suppressor gene in sporadic follicular thyroid tumors. Genes Chromosom. Cancer 1998, 23, 239–243.
- Hsieh, M.C.; Lin, S.F.; Shin, S.J.; Liu, T.C.; Chang, J.G.; Lee, J.P. Mutation analysis of PTEN/MMAC 1 in sporadic thyroid tumors. Kaohsiung J. Med. Sci. 2000, 16, 9–12.
- Hou, P.; Ji, M.; Xing, M. Association of PTEN gene methylation with genetic alterations in the phosphatidylinositol 3-kinase/AKT signaling pathway in thyroid tumors. Cancer 2008, 113, 2440–2447.
- Alvarez-Nunez, F.; Bussaglia, E.; Mauricio, D.; Ybarra, J.; Vilar, M.; Lerma, E.; de Leiva, A.; Matias-Guiu, X. PTEN promoter methylation in sporadic thyroid carcinomas. Thyroid 2006, 16, 17–23.
- Pringle, D.R.; Vasko, V.V.; Yu, L.; Manchanda, P.K.; Lee, A.A.; Zhang, X.; Kirschner, J.M.; Parlow, A.F.; Saji, M.; Jarjoura, D.; et al. Follicular thyroid cancers demonstrate dual activation of PKA and mTOR as modeled by thyroid-specific deletion of Prkar1a and Pten in mice. J. Clin. Endocrinol. Metab. 2014, 99, E804–E812.
- Bruni, P.; Boccia, A.; Baldassarre, G.; Trapasso, F.; Santoro, M.; Chiappetta, G.; Fusco, A.; Viglietto, G. PTEN expression is reduced in a subset of sporadic thyroid carcinomas: Evidence that PTEN-growth suppressing activity in thyroid cancer cells mediated by p27kip1. Oncogene 2000, 19, 3146–3155.
- Beg, S.; Siraj, A.K.; Jehan, Z.; Prabakaran, S.; Al-Sobhi, S.S.; Al-Dawish, M.; Al-Dayel, F.; Al-Kuraya, K.S. PTEN loss is associated with follicular variant of Middle Eastern papillary thyroid carcinoma. Br. J. Cancer 2015, 112, 1938–1943.
- Yu, W.; Ni, Y.; Saji, M.; Ringel, M.D.; Jaini, R.; Eng, C. Cowden syndrome-associated germline succinate dehydrogenase complex subunit D (SDHD) variants cause PTEN-mediated down-regulation of autophagy in thyroid cancer cells. Hum. Mol. Genet. 2017, 26, 1365–1375.
- Frisk, T.; Foukakis, T.; Dwight, T.; Lundberg, J.; Hoog, A.; Wallin, G.; Eng, C.; Zedenius, J.; Larsson, C. Silencing of the PTEN tumor-suppressor gene in anaplastic thyroid cancer. Genes Chromosom. Cancer 2002, 35, 74–80.
- Groden, J.; Thliveris, A.; Samowitz, W.; Carlson, M.; Gelbert, L.; Albertsen, H.; Joslyn, G.; Stevens, J.; Spirio, L.; Robertson, M.; et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell 1991, 66, 589–600.
- Beroud, C.; Soussi, T. APC gene: Database of germline and somatic mutations in human tumors and cell lines. Nucleic Acids Res. 1996, 24, 121–124.
- Crail, H.W. Multiple primary malignancies arising in the rectum, brain, and thyroid; report of a case. US Nav. Med. Bull. 1949, 49, 123–128.
- Bulow, C.; Bulow, S. Is screening for thyroid carcinoma indicated in familial adenomatous polyposis? The Leeds Castle Polyposis Group. Int. J. Colorectal Dis. 1997, 12, 240–242.
- Cetta, F.; Montalto, G.; Gori, M.; Curia, M.C.; Cama, A.; Olschwang, S. Germline mutations of the APC gene in patients with familial adenomatous polyposis-associated thyroid carcinoma: Results from a European cooperative study. J. Clin. Endocrinol. Metab. 2000, 85, 286–292.
- Groen, E.J.; Roos, A.; Muntinghe, F.L.; Enting, R.H.; de Vries, J.; Kleibeuker, J.H.; Witjes, M.J.; Links, T.P.; van Beek, A.P. Extra-intestinal manifestations of familial adenomatous polyposis. Ann. Surg. Oncol. 2008, 15, 2439–2450.
- Martayan, A.; Sanchez-Mete, L.; Baldelli, R.; Falvo, E.; Barnabei, A.; Conti, L.; Giacomini, P.; Appetecchia, M.; Stigliano, V. Gene variants associated to malignant thyroid disease in familial adenomatous polyposis: A novel APC germline mutation. J. Endocrinol. Investig. 2010, 33, 603–606.
- Zeki, K.; Spambalg, D.; Sharifi, N.; Gonsky, R.; Fagin, J.A. Mutations of the adenomatous polyposis coli gene in sporadic thyroid neoplasms. J. Clin. Endocrinol. Metab. 1994, 79, 1317–1321.
- Cetta, F.; Chiappetta, G.; Melillo, R.M.; Petracci, M.; Montalto, G.; Santoro, M.; Fusco, A. The ret/ptc1 oncogene is activated in familial adenomatous polyposis-associated thyroid papillary carcinomas. J. Clin. Endocrinol. Metab. 1998, 83, 1003–1006.
- Uchino, S.; Noguchi, S.; Yamashita, H.; Yamashita, H.; Watanabe, S.; Ogawa, T.; Tsuno, A.; Murakami, A.; Miyauchi, A. Mutational analysis of the APC gene in cribriform-morula variant of papillary thyroid carcinoma. World J. Surg. 2006, 30, 775–779.
- Cetta, F.; Olschwang, S.; Petracci, M.; Montalto, G.; Baldi, C.; Zuckermann, M.; Mariani Costantini, R.; Fusco, A. Genetic alterations in thyroid carcinoma associated with familial adenomatous polyposis: Clinical implications and suggestions for early detection. World J. Surg. 1998, 22, 1231–1236.
- Truta, B.; Allen, B.A.; Conrad, P.G.; Kim, Y.S.; Berk, T.; Gallinger, S.; Bapat, B.; Terdiman, J.P.; Sleisenger, M.H. Genotype and phenotype of patients with both familial adenomatous polyposis and thyroid carcinoma. Fam. Cancer 2003, 2, 95–99.
- Septer, S.; Slowik, V.; Morgan, R.; Dai, H.; Attard, T. Thyroid cancer complicating familial adenomatous polyposis: Mutation spectrum of at-risk individuals. Hered. Cancer Clin. Pract. 2013, 11, 13.
- Han, S.H.; Ryu, J.S.; Kim, Y.J.; Cho, H.I.; Yang, Y.H.; Lee, K.R. Mutation analysis of the APC gene in unrelated Korean patients with FAP: Four novel mutations with unusual phenotype. Fam. Cancer 2011, 10, 21–26.
- Zhang, X.; Li, M.; Zuo, K.; Li, D.; Ye, M.; Ding, L.; Cai, H.; Fu, D.; Fan, Y.; Lv, Z. Upregulated miR-155 in papillary thyroid carcinoma promotes tumor growth by targeting APC and activating Wnt/beta-catenin signaling. J. Clin. Endocrinol. Metab. 2013, 98, E1305–E1313.
- Rubinfeld, B.; Albert, I.; Porfiri, E.; Munemitsu, S.; Polakis, P. Loss of beta-catenin regulation by the APC tumor suppressor protein correlates with loss of structure due to common somatic mutations of the gene. Cancer Res. 1997, 57, 4624–4630.
- Kumamoto, K.; Ishida, H.; Ohsawa, T.; Ishibashi, K.; Ushiama, M.; Yoshida, T.; Iwama, T. Germline and somatic mutations of the APC gene in papillary thyroid carcinoma associated with familial adenomatous polyposis: Analysis of three cases and a review of the literature. Oncol. Lett. 2015, 10, 2239–2243.
- Xing, M. RASAL1 in thyroid cancer: Promise from a new friend. J. Clin. Endocrinol. Metab. 2014, 99, 3619–3621.
- Liu, D.; Yang, C.; Bojdani, E.; Murugan, A.K.; Xing, M. Identification of RASAL1 as a major tumor suppressor gene in thyroid cancer. J. Natl. Cancer Inst. Monogr. 2013, 105, 1617–1627.
- Ngeow, J.; Ni, Y.; Tohme, R.; Song Chen, F.; Bebek, G.; Eng, C. Germline alterations in RASAL1 in Cowden syndrome patients presenting with follicular thyroid cancer and in individuals with apparently sporadic epithelial thyroid cancer. J. Clin. Endocrinol. Metab. 2014, 99, E1316–E1321.
- Lyssikatos, C.; Quezado, M.M.; Faucz, F.R.; Angelousi, A.; Nasiri-Ansari, N.; Stratakis, C.A.; Kassi, E. A rare case of medullary thyroid cancer, mesothelioma and meningioma, due to APC and RASAL1 mutations. In Proceedings of the 19th European Congress of Endocrinology, Lisbon, Portugal, 20–23 May 2017.
- Angelousi, A.; Settas, N.; Faucz, F.R.; Lyssikatos, C.; Quezado, M.; Nasiri-Ansari, N.; Stratakis, C.A.; Kassi, E. Medullary thyroid cancer, leukemia, mesothelioma and meningioma associated with germline APC and RASAL1 variants: A new syndrome? Hormones 2017, 16, 423–428.
- Jeon, M.J.; Chun, S.M.; Kim, D.; Kwon, H.; Jang, E.K.; Kim, T.Y.; Kim, W.B.; Shong, Y.K.; Jang, S.J.; Song, D.E.; et al. Genomic Alterations of Anaplastic Thyroid Carcinoma Detected by Targeted Massive Parallel Sequencing in a BRAF(V600E) Mutation-Prevalent Area. Thyroid 2016, 26, 683–690.
- Preto, A.; Reis-Filho, J.S.; Ricardo, S.; Soares, P. P63 expression in papillary and anaplastic carcinomas of the thyroid gland: Lack of an oncogenetic role in tumorigenesis and progression. Pathol. Res. Pract. 2002, 198, 449–454.
- Reis-Filho, J.S.; Preto, A.; Soares, P.; Ricardo, S.; Cameselle-Teijeiro, J.; Sobrinho-Simoes, M. p63 expression in solid cell nests of the thyroid: Further evidence for a stem cell origin. Mod. Pathol. 2003, 16, 43–48.
- Bonzanini, M.; Amadori, P.L.; Sagramoso, C.; Dalla Palma, P. Expression of cytokeratin 19 and protein p63 in fine needle aspiration biopsy of papillary thyroid carcinoma. Acta Cytol. 2008, 52, 541–548.
- Malaguarnera, R.; Mandarino, A.; Mazzon, E.; Vella, V.; Gangemi, P.; Vancheri, C.; Vigneri, P.; Aloisi, A.; Vigneri, R.; Frasca, F. The p53-homologue p63 may promote thyroid cancer progression. Endocr. Relat. Cancer 2005, 12, 953–971.
- Ferru, A.; Denis, S.; Guilhot, J.; Gibelin, H.; Tourani, J.M.; Kraimps, J.L.; Larsen, C.J.; Karayan-Tapon, L. Expression of TAp73 and DeltaNp73 isoform transcripts in thyroid tumours. Eur. J. Surg. Oncol. 2006, 32, 228–230.
- Frasca, F.; Vella, V.; Aloisi, A.; Mandarino, A.; Mazzon, E.; Vigneri, R.; Vigneri, P. p73 tumor-suppressor activity is impaired in human thyroid cancer. Cancer Res. 2003, 63, 5829–5837.
- Ito, Y.; Uramoto, H.; Funa, K.; Yoshida, H.; Jikuzono, T.; Asahi, S.; Higashiyama, T.; Tomoda, C.; Takamura, Y.; Miya, A.; et al. Delta Np73 expression in thyroid neoplasms originating from follicular cells. Pathology 2006, 38, 205–209.
- Puppin, C.; Fabbro, D.; Dima, M.; Di Loreto, C.; Puxeddu, E.; Filetti, S.; Russo, D.; Damante, G. High periostin expression correlates with aggressiveness in papillary thyroid carcinomas. J. Endocrinol. 2008, 197, 401–408.
- Puppin, C.; Passon, N.; Frasca, F.; Vigneri, R.; Tomay, F.; Tomaciello, S.; Damante, G. In thyroid cancer cell lines expression of periostin gene is controlled by p73 and is not related to epigenetic marks of active transcription. Cell. Oncol. 2011, 34, 131–140.
- Vella, V.; Puppin, C.; Damante, G.; Vigneri, R.; Sanfilippo, M.; Vigneri, P.; Tell, G.; Frasca, F. DeltaNp73alpha inhibits PTEN expression in thyroid cancer cells. Int. J. Cancer 2009, 124, 2539–2548.
- Figge, J.; Bakst, G.; Weisheit, D.; Solis, O.; Ross, J.S. Image analysis quantitation of immunoreactive retinoblastoma protein in human thyroid neoplasms with a streptavidin-biotin-peroxidase staining technique. Am. J. Pathol. 1991, 139, 1213–1219.
- Zou, M.; Shi, Y.; Farid, N.R.; Al-Sedairy, S.T. Inverse association between cyclin D1 overexpression and retinoblastoma gene mutation in thyroid carcinomas. Endocrine 1998, 8, 61–64.
- Holm, R.; Nesland, J.M. Retinoblastoma and p53 tumour suppressor gene protein expression in carcinomas of the thyroid gland. J. Pathol. 1994, 172, 267–272.
- Van Veelen, W.; van Gasteren, C.J.; Acton, D.S.; Franklin, D.S.; Berger, R.; Lips, C.J.; Hoppener, J.W. Synergistic effect of oncogenic RET and loss of p18 on medullary thyroid carcinoma development. Cancer Res. 2008, 68, 1329–1337.
- Cote, G.J.; Grubbs, E.G.; Hofmann, M.C. Thyroid C-Cell Biology and Oncogenic Transformation. Recent Results Cancer Res. 2015, 204, 1–39.
- Pozo, K.; Hillmann, A.; Augustyn, A.; Plattner, F.; Hai, T.; Singh, T.; Ramezani, S.; Sun, X.; Pfragner, R.; Minna, J.D.; et al. Differential expression of cell cycle regulators in CDK5-dependent medullary thyroid carcinoma tumorigenesis. Oncotarget 2015, 6, 12080–12093.
- Song, H.; Lin, C.; Yao, E.; Zhang, K.; Li, X.; Wu, Q.; Chuang, P.T. Selective Ablation of Tumor Suppressors in Parafollicular C Cells Elicits Medullary Thyroid Carcinoma. J. Biol. Chem. 2017, 292, 3888–3899.
- Valenciaga, A.; Grubbs, E.G.; Porter, K.; Wakely, P.E., Jr.; Williams, M.D.; Cote, G.J.; Vasko, V.V.; Saji, M.; Ringel, M.D. Reduced Retinoblastoma Protein Expression Is Associated with Decreased Patient Survival in Medullary Thyroid Cancer. Thyroid 2017, 27, 1523–1533.
- Brzezinski, J.; Migodzinski, A.; Toczek, A.; Tazbir, J.; Dedecjus, M. Patterns of cyclin E, retinoblastoma protein, and p21Cip1/WAF1 immunostaining in the oncogenesis of papillary thyroid carcinoma. Clin. Cancer Res. 2005, 11, 1037–1043.
- Skalhegg, B.S.; Tasken, K. Specificity in the cAMP/PKA signaling pathway. Differential expression, regulation, and subcellular localization of subunits of PKA. Front. Biosci. 2000, 5, D678–D693.
- Gu, T.; Zhang, Z.; Wang, J.; Guo, J.; Shen, W.H.; Yin, Y. CREB is a novel nuclear target of PTEN phosphatase. Cancer Res. 2011, 71, 2821–2825.
- Massimi, M.; Cardarelli, S.; Galli, F.; Giardi, M.F.; Ragusa, F.; Panera, N.; Cinque, B.; Cifone, M.G.; Biagioni, S.; Giorgi, M. Increase of Intracellular Cyclic AMP by PDE4 Inhibitors Affects HepG2 Cell Cycle Progression and Survival. J. Cell. Biochem. 2017, 118, 1401–1411.
- Yasuda, M.T.; Sakakibara, H.; Shimoi, K. Estrogen- and stress-induced DNA damage in breast cancer and chemoprevention with dietary flavonoid. Genes Environ. 2017, 39, 10.
- Kirschner, L.S.; Carney, J.A.; Pack, S.D.; Taymans, S.E.; Giatzakis, C.; Cho, Y.S.; Cho-Chung, Y.S.; Stratakis, C.A. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat. Genet. 2000, 26, 89–92.
- Sandrini, F.; Matyakhina, L.; Sarlis, N.J.; Kirschner, L.S.; Farmakidis, C.; Gimm, O.; Stratakis, C.A. Regulatory subunit type I-alpha of protein kinase A (PRKAR1A): A tumor-suppressor gene for sporadic thyroid cancer. Genes Chromosomes Cancer 2002, 35, 182–192.
- Pringle, D.R.; Yin, Z.; Lee, A.A.; Manchanda, P.K.; Yu, L.; Parlow, A.F.; Jarjoura, D.; La Perle, K.M.; Kirschner, L.S. Thyroid-specific ablation of the Carney complex gene, PRKAR1A, results in hyperthyroidism and follicular thyroid cancer. Endocr. Relat. Cancer 2012, 19, 435–446.
- Matsuoka, S.; Huang, M.; Elledge, S.J. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 1998, 282, 1893–1897.
- Bartkova, J.; Horejsi, Z.; Koed, K.; Kramer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870.
- Cybulski, C.; Huzarski, T.; Gorski, B.; Masojc, B.; Mierzejewski, M.; Debniak, T.; Gliniewicz, B.; Matyjasik, J.; Zlowocka, E.; Kurzawski, G.; et al. A novel founder CHEK2 mutation is associated with increased prostate cancer risk. Cancer Res. 2004, 64, 2677–2679.
- Kilpivaara, O.; Vahteristo, P.; Falck, J.; Syrjakoski, K.; Eerola, H.; Easton, D.; Bartkova, J.; Lukas, J.; Heikkila, P.; Aittomaki, K.; et al. CHEK2 variant I157T may be associated with increased breast cancer risk. Int. J. Cancer 2004, 111, 543–547.
- Cybulski, C.; Gorski, B.; Huzarski, T.; Masojc, B.; Mierzejewski, M.; Debniak, T.; Teodorczyk, U.; Byrski, T.; Gronwald, J.; Matyjasik, J.; et al. CHEK2 is a multiorgan cancer susceptibility gene. Am. J. Hum. Genet. 2004, 75, 1131–1135.
- Wojcicka, A.; Czetwertynska, M.; Swierniak, M.; Dlugosinska, J.; Maciag, M.; Czajka, A.; Dymecka, K.; Kubiak, A.; Kot, A.; Ploski, R.; et al. Variants in the ATM-CHEK2-BRCA1 axis determine genetic predisposition and clinical presentation of papillary thyroid carcinoma. Genes Chromosomes Cancer 2014, 53, 516–523.
- Fayaz, S.; Fard-Esfahani, P.; Torbati, P.M. Lack of CHEK2 gene mutations in differentiated thyroid carcinoma patients using high resolution melting analysis. Asian Pac. J. Cancer Prev. 2014, 15, 5019–5022.
- Kaczmarek-Rys, M.; Ziemnicka, K.; Hryhorowicz, S.T.; Gorczak, K.; Hoppe-Golebiewska, J.; Skrzypczak-Zielinska, M.; Tomys, M.; Golab, M.; Szkudlarek, M.; Budny, B.; et al. The c.470 T > C CHEK2 missense variant increases the risk of differentiated thyroid carcinoma in the Great Poland population. Hered. Cancer Clin. Pract 2015, 13, 8.
- Swierniak, M.; Pfeifer, A.; Stokowy, T.; Rusinek, D.; Chekan, M.; Lange, D.; Krajewska, J.; Oczko-Wojciechowska, M.; Czarniecka, A.; Jarzab, M.; et al. Somatic mutation profiling of follicular thyroid cancer by next generation sequencing. Mol. Cell. Endocrinol. 2016, 433, 130–137.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Revisions:
3 times
(View History)
Update Date:
31 May 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No