 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Vladimir Chubanov | + 1434 word(s) | 1434 | 2020-09-30 04:25:04 | | | |
| 2 | Nicole Yin | Meta information modification | 1434 | 2020-10-12 04:45:26 | | | | |
| 3 | Nicole Yin | Meta information modification | 1434 | 2020-11-02 05:14:08 | | |
Video Upload Options
The transient receptor potential cation channel, subfamily M, member 7 (TRPM7) is a ubiquitously expressed membrane protein, which forms a cation channel linked to a cytosolic protein kinase.
1. Introduction
TRPM7 has been cloned and functionally characterized two decades ago[1][2][3]. Since then, extensive investigations have been conducted to clarify the molecular and organismal aspects of the TRPM7 function[4]. Genetic inactivation of TRPM7 in animal models uncovered the critical role of TRPM7 in early embryonic development, immune responses, and the organismal balance of Zn2+, Mg2+, and Ca2+. TRPM7 emerged as a new therapeutic target because malfunctions of TRPM7 have been associated with anoxic neuronal death, tissue fibrosis, tumour progression, and giant platelet disorder.
2. Functional Characteristics and Physiological Roles of TRPM7
TRPM7 encodes a bi-functional protein comprising a TRP-type transmembrane channel unit fused to a C-terminal α-type serine/threonine-protein kinase domain[1][2][3]. Similarly to other TRP channels, the channel-coding segment of TRPM7 comprises six transmembrane helixes with a channel pore-forming sequence located between the fifth and six helices (Figure 1A,B). Four TRPM7 proteins assemble in a symmetric channel complex (Figure 1C)[1][2][3]. Hence, one TRPM7 channel moiety is linked to four cytosolic kinase domains. Among other known channels and kinases, only TRPM7 and its homologous protein TRPM6 are known as channels covalently fused to protein kinase domains[5][6][7][8]. The crystal structure of the C-terminal TRPM7 domain revealed the three-dimensional packing of the catalytic domain of the kinase[9]. More recently, cryo-electron microscopy of the truncated TRPM7 protein (lacking the kinase domain) clarified the role of distinct amino acid residues for the tetrameric assembly of the channel segment (Figure 1B,C)[10]. However, the positioning of the kinase and channel units relative to each other in the full-length TRPM7 protein, as well as distinct rearrangements in TRPM7 folding during channel gating, remain unknown[11].
In pioneering patch-clamp experiments, endogenous TRPM7 currents were referred to as magnesium nucleotide-regulated metal ion currents (MagNuM)[1][12] and magnesium-inhibited cation currents (MIC)[13], and were later called TRPM7-like or TRPM7 currents[14][15][16][17][18][19][20]. Such native TRPM7 currents were monitored in a large variety of primary isolated cells and stable cell lines[14][15][16][17][18][19][20]. In accord with biophysical experiments, TRPM7 transcripts were found to be abundantly present in all native tissues examined[14][15][16][17][18][19][20]. TRPM7 was defined as a constitutively active cation channel highly selective for divalent cations such as Zn2+, Ca2+ and Mg2+[1][2][3]. Among other factors, cytosolic magnesium (free Mg2+ or in complex Mg·ATP) and the plasma membrane phospholipid phosphatidylinositol-4,5-bisphosphate (PIP2) were discovered as prime physiological regulators of TRPM7[1][2][3][21]. While intracellular Mg2+ or Mg·ATP directly act as negative regulators of the channel, receptor-dependent phospholipase C activation, and resultant PIP2 depletion indirectly result in TRPM7 inactivation[1][2][3][21].
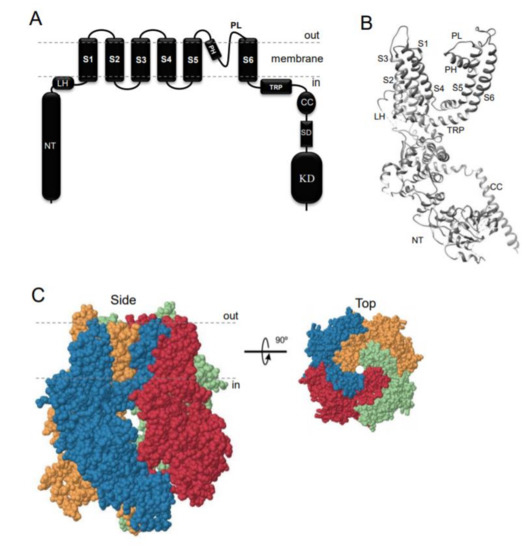
Figure 1. Domain topology and tetrameric assembly of the kinase-coupled channel TRPM7. (A) TRPM7 comprises a large cytosolic N-terminal domain (NT), a linker-helical domain (LH), six transmembrane helices (S1-S6), a pore-forming pore helix (PH) and loop (PL), a transient receptor potential domain (TRP), a coiled-coil domain (CC), a kinase substrate domain (SD) and a kinase domain (KD). (B) Ribbon diagram of a single TRPM7 channel subunit produced from 6BWD using UCSF Chimera (www.cgl.ucsf.edu). (C) Tetrameric TRPM7 channel complex (6BWD) Four channel subunits of TRPM7 are labelled by different colours and shown from the side and top views.
Genetic disruption of TRPM7 in cultured cells revealed that the TRPM7 channel is key to the homeostatic balance of divalent cations including Zn2+, Mg2+ and Ca2+[7][22][23][24][25][26], cell motility[27][28][29][30][31][32][33][34], proliferation[1][23][24][35][36][37], differentiation[38][39], Ca2+ signaling events[40][41] and an ever growing number of other cellular processes[14][15][16][17][18][19][20]. Pathophysiological implications of TRPM7 are widespread and include anoxic neuronal death[42], hypertension[43][44], neurodegenerative disorders[45][46], tissue fibrosis[47][48][49][50], tumour growth/progression[51][52][53][54][55][56][57][58] and abnormal[47][48][49][50] immune responses[59]. Genetic association studies in humans revealed that point mutations in the TRPM7 gene cause a giant platelet disorder (macrothrombocytopenia)[60]. Experiments with mice currying a global or tissue-specific null mutation in the Trpm7 locus showed that TRPM7 is required for early embryonic development[22][61][62][63][64], thymopoiesis[61], morphogenesis of the kidney[63], cardiac rhythmicity and repolarization[65] [65], systemic homeostasis of Zn2+, Mg2+ and Ca2+[22][66], thrombopoiesis[60], and mast cell degranulation[67].
The list of phosphorylation substrates of the TRPM7 kinase is extensive and surprisingly heterogeneous in terms of possible biochemical pathways affected. Thus, TRPM7 kinase can phosphorylate TRPM6[68], annexin A1[69], myosin II isoforms[70], eukaryotic elongation factor-2 kinase (eEF2-k)[71], tropomodulin[72], phospholipase C gamma 2 (PLCγ2)[73], stromal interaction molecule 2 (STIM2)[25], Mothers against decapentaplegic homolog 2 (SMAD2)[59], and Ras homolog family member A (RhoA)[74]. Furthermore, multiple serine/threonine residues positioned in a ‘substrate’ segment of TRPM7 are autophosphorylation targets of the kinase domain[20][75][76][77] . In immune cells, the TRPM7 kinase domain can be cleaved from the channel complex by caspases during Fas-receptor stimulation[35]. Another study reported that the cleaved TRPM7 kinase can be detected in several cell lines and that the released kinase is able to translocate into the cell nucleus to phosphorylate histones[78]. The in vivo relevance of these reactions remains to be verified, because, unlike to the mouse strains with the Trpm7 null mutation, animals carrying the ‘kinase-dead’ point mutation were found to be fertile, and displayed normal pre- and postnatal development, if maintained under regular conditions[59][66][79][80].
3. Drug-like Modulators the Channel and Kinase Activity of TRPM7
In light of the bi-functional nature of TRPM7, there is a growing demand for reliable drug-like molecules allowing for selective and distinct modulation of its channel and kinase moieties. Initially, agents acting as unspecific channel inhibitors, such as spermine[13], ruthenium red[81], trivalent cations[82], SKF-96365[13] and 2-aminoethyl diphenylborinate (2-APB)[83], were used to block the TRPM7 channel. Subsequently, several drug-like molecules were reported as inhibitors of the TRPM7 channel effective only in a high µM range, such as nafamostat[84], carvacrol[85][86][87][88][89], 5-lipoxygenase inhibitors (NDGA, AA861 and MK886)[90][91][92][93], midazolam[94][95], ginsenoside Rg3[96], ginsenoside-Rd[97][98], aripiprazole[99] and coomassie brilliant blue G-250 (BBG)[100]. Our laboratory identified several additional inhibitors of the TRPM7 channel such as quinine, CyPPA, dequalinium, SKA31, and UCL1684[101].
In contrast to later molecules, Waixenicin A, FTY720 and NS8593 were able to suppress TRPM7 currents when applied at low µM concentrations. Subsequently, these reagents were often used to probe the cellular role of TRPM7[102][103]. Waixenicin A is a natural terpenoid isolated from the soft coral Sarcothelia edmondsoni, and inactivates the TRPM7 channel in an Mg2+ dependent manner with an IC50 of 7 µM[37]. FTY720 (synthetic homolog of sphingosine) inhibited TRPM7 currents with an IC50 0.7 µM[104]. Our laboratory has shown that the small synthetic molecule N-[(1R)-1,2,3,4-tetrahydronaphthalen-1-yl]-1H-benzimidazol-2-amine (NS8593, Figure 2) suppresses TRPM7 currents in an Mg2+-dependent fashion with an IC50 of 1.6 µM[101].
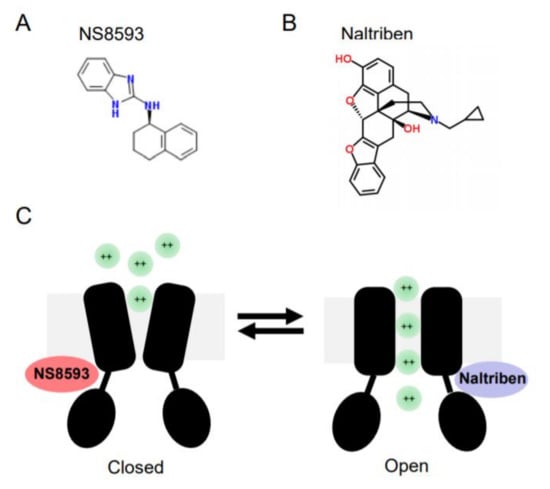
Figure 2. Chemical structures and mode of action of NS8593 and naltriben on the TRPM7 channel. (A) NS8593 chemical structure. (B) Naltriben chemical structure. (C)The TRPM7 channel is depicted in the closed and open states. NS8593 acts as negative gating modulator of the channel, whereas naltiben triggers opening of TRPM7 and influx of divalent cations (green balls) into the cell.
In a follow-up screen, our group has discovered the first small drug-like molecules functioning as TRPM7 channel agonists[105]. In particular, we found that twenty drug-like compounds with different structural backbones can stimulate TRPM7 currents[105][106]. Among them, naltriben (Figure 2) and mibefradil were characterized more in detail, and nowadays both compounds are frequently used by other TRPM7 investigators, often in combination with the TRPM7 inhibitors mentioned before[102][103]. Of note, naltriben is able to activate the TRPM7 channel both in the presence of physiological concentrations of cytosolic Mg2+ and after PIP2 depletion with an EC50 of 20 µM[105]. Hence, we defined naltriben as a positive gating modulator of the TRPM7 channel[105]. Unlike naltriben, mibefradil-mediated activation of TRPM7 was highly dependent on intracellular Mg2+ levels[106]. Accordingly, we suggested that at least two distinct types of TRPM7 activators exist, referred to as type 1 (acting independently of Mg2+) and type 2 (Mg2+-dependent agonists)[106].
Overall, the pharmacological toolkit suitable for the assessment of the TRPM7 kinase remains underdeveloped, and currently, it is limited to only one compound, TG100-115. TG100-115 was initially introduced as an inhibitor of phosphoinositide 3-kinases[107]. However, Davis et al. [108] found that TG100-115 is also able to inactivate the purified kinase domain of TRPM6 with an IC50 of 8 nM[108] [108]. We also found that TG100-115 efficiently inactivates TRPM6 kinase in living cells[21]. Besides, Song et al.[109] examined the effects of TG100-115 on the TRPM7 kinase and reported that this reagent inhibits the TRPM7 kinase with an IC50 of 2 µM. Finally, it is worth mentioning that in our hands, neither NS8593 nor naltriben directly affected the kinase activity of TRPM7 (unpublished observations). However, we cannot rigorously exclude that in specific experimental settings, these compounds may modulate the kinase moiety indirectly, for instance, subsequent to altered uptake of divalent cations by the channel domain of TRPM7.
References
- Nadler, M.J.; Hermosura, M.C.; Inabe, K.; Perraud, A.L.; Zhu, Q.; Stokes, A.J.; Kurosaki, T.; Kinet, J.P.; Penner, R.; Scharenberg, A.M.; et al. Ltrpc7 is a mg.Atp-regulated divalent cation channel required for cell viability. Nature 2001, 411, 590–595.
- Runnels, L.W.; Yue, L.; Clapham, D.E. Trp-plik, a bifunctional protein with kinase and ion channel activities. Science 2001, 291, 1043–1047.
- Ryazanov, A.G.; Ward, M.D.; Mendola, C.E.; Pavur, K.S.; Dorovkov, M.V.; Wiedmann, M.; Erdjument-Bromage, H.; Tempst, P.; Parmer, T.G.; Prostko, C.R.; et al. Identification of a new class of protein kinases represented by eukaryotic elongation factor-2 kinase. Proc. Natl. Acad. Sci. USA 1997, 94, 4884–4889.
- Shiqi Zhang; Dongyi Zhao; Wanying Jia; Yuting Wang; Hongyue Liang; Lei Liu; Wuyang Wang; Zhiyi Yu; Feng Guo; A bibliometric analysis and review of recent researches on TRPM7. Channels 2020, 14, 203-215, 10.1080/19336950.2020.1788355.
- Chubanov, V.; Ferioli, S.; Wisnowsky, A.; Simmons, D.G.; Leitzinger, C.; Einer, C.; Jonas, W.; Shymkiv, Y.; Bartsch, H.; Braun, A.; et al. Epithelial magnesium transport by trpm6 is essential for prenatal development and adult survival. Elife 2016, 5, e20914.
- Chubanov, V.; Gudermann, T. Trpm6. Handb. Exp. Pharmacol. 2014, 222, 503–520.
- Chubanov, V.; Waldegger, S.; Mederos y Schnitzler, M.; Vitzthum, H.; Sassen, M.C.; Seyberth, H.W.; Konrad, M.; Gudermann, T. Disruption of trpm6/trpm7 complex formation by a mutation in the trpm6 gene causes hypomagnesemia with secondary hypocalcemia. Proc. Natl. Acad. Sci. USA 2004, 101, 2894–2899.
- Mederos y Schnitzler, M.; Waring, J.; Gudermann, T.; Chubanov, V. Evolutionary determinants of divergent calcium selectivity of trpm channels. FASEB J. 2008, 22, 1540–1551.
- Hiroto Yamaguchi; Masayuki Matsushita; Angus C. Nairn; J Kuriyan; Crystal Structure of the Atypical Protein Kinase Domain of a TRP Channel with Phosphotransferase Activity. Molecular Cell 2001, 7, 1047-1057, 10.1016/s1097-2765(01)00256-8.
- Jingjing Duan; Zongli Li; Jian Li; Raymond E. Hulse; Ana Santa-Cruz; William C. Valinsky; Sunday A. Abiria; Grigory Krapivinsky; Qisheng Wang; David E. Clapham; et al. Structure of the mammalian TRPM7, a magnesium channel required during embryonic development.. Proceedings of the National Academy of Sciences 2018, 115, E8201-E8210, 10.1073/pnas.1810719115.
- Vladimir Chubanov; Lorenz Mittermeier; Thomas Gudermann; TRPM7 reflected in Cryo-EMirror.. Cell Calcium 2018, 76, 129-131, 10.1016/j.ceca.2018.11.004.
- Meredith C Hermosura; Mahealani K Monteilh-Zoller; Andrew M. Scharenberg; Reinhold Penner; Andrea Fleig; Dissociation of the store-operated calcium current I(CRAC) and the Mg-nucleotide-regulated metal ion current MagNuM.. The Journal of Physiology 2002, 539, 445–458.
- J. Ashot Kozak; Hubert H. Kerschbaum; Michael D. Cahalan; Distinct Properties of CRAC and MIC Channels in RBL Cells. Journal of General Physiology 2002, 120, 221-235, 10.1085/jgp.20028601.
- Chubanov, V.; Mittermeier, L.; Gudermann, T. Role of kinase-coupled trp channels in mineral homeostasis. Pharmacol. Ther 2018, 184, 159–176.
- Fleig, A.; Chubanov, V. Trpm7. Handb Exp. Pharmacol. 2014, 222, 521–546.
- Abumaria, N.; Li, W.; Clarkson, A.N. Role of the chanzyme trpm7 in the nervous system in health and disease. Cell Mol. Life Sci 2019, 76, 3301–3310.
- Zou, Z.G.; Rios, F.J.; Montezano, A.C.; Touyz, R.M. Trpm7, magnesium, and signaling. Int J. Mol. Sci. 2019, 20, 1877.
- Runnels, L.W.; Komiya, Y. Trpm6 and trpm7: Novel players in cell intercalation during vertebrate embryonic development. Dev. Dyn 2020.
- Nadolni, W.; Zierler, S. The channel-kinase trpm7 as novel regulator of immune system homeostasis. Cells 2018, 7, 109.
- Bates-Withers, C.; Sah, R.; Clapham, D.E. Trpm7, the mg(2+) inhibited channel and kinase. Adv. Exp. Med. Biol. 2011, 704, 173–183.
- Silvia Ferioli; Susanna Zierler; Joanna Zaißerer; Johann Schredelseker; Thomas Gudermann; Vladimir Chubanov; TRPM6 and TRPM7 differentially contribute to the relief of heteromeric TRPM6/7 channels from inhibition by cytosolic Mg2+ and Mg·ATP. Scientific Reports 2017, 7, 1-19, 10.1038/s41598-017-08144-1.
- Ryazanova, L.V.; Rondon, L.J.; Zierler, S.; Hu, Z.; Galli, J.; Yamaguchi, T.P.; Mazur, A.; Fleig, A.; Ryazanov, A.G. Trpm7 is essential for mg(2+) homeostasis in mammals. Nat. Commun. 2010, 1, 109.
- Sahni, J.; Scharenberg, A.M. Trpm7 ion channels are required for sustained phosphoinositide 3-kinase signaling in lymphocytes. Cell Metab. 2008, 8, 84–93.
- Schmitz, C.; Perraud, A.L.; Johnson, C.O.; Inabe, K.; Smith, M.K.; Penner, R.; Kurosaki, T.; Fleig, A.; Scharenberg, A.M. Regulation of vertebrate cellular mg2+ homeostasis by trpm7. Cell 2003, 114, 191–200.
- Faouzi, M.; Kilch, T.; Horgen, F.D.; Fleig, A.; Penner, R. The trpm7 channel kinase regulates store-operated calcium entry. J. Physiol. 2017, 595, 3165–3180.
- Abiria, S.A.; Krapivinsky, G.; Sah, R.; Santa-Cruz, A.G.; Chaudhuri, D.; Zhang, J.; Adstamongkonkul, P.; DeCaen, P.G.; Clapham, D.E. Trpm7 senses oxidative stress to release zn(2+) from unique intracellular vesicles. Proc. Natl. Acad. Sci. USA 2017, 114, E6079–E6088.
- Su, L.T.; Agapito, M.A.; Li, M.; Simonson, W.T.; Huttenlocher, A.; Habas, R.; Yue, L.; Runnels, L.W. Trpm7 regulates cell adhesion by controlling the calcium-dependent protease calpain. J. Biol. Chem. 2006, 281, 11260–11270.
- Wei, C.; Wang, X.; Chen, M.; Ouyang, K.; Song, L.S.; Cheng, H. Calcium flickers steer cell migration. Nature 2009, 457, 901–905.
- Clark, K.; Langeslag, M.; van Leeuwen, B.; Ran, L.; Ryazanov, A.G.; Figdor, C.G.; Moolenaar, W.H.; Jalink, K.; van Leeuwen, F.N. Trpm7, a novel regulator of actomyosin contractility and cell adhesion. EMBO J. 2006, 25, 290–301.
- Meng, X.; Cai, C.; Wu, J.; Cai, S.; Ye, C.; Chen, H.; Yang, Z.; Zeng, H.; Shen, Q.; Zou, F. Trpm7 mediates breast cancer cell migration and invasion through the mapk pathway. Cancer Lett. 2013, 333, 96–102.
- Siddiqui, T.A.; Lively, S.; Vincent, C.; Schlichter, L.C. Regulation of podosome formation, microglial migration and invasion by ca(2+)-signaling molecules expressed in podosomes. J. Neuroinflamm. 2012, 9, 250.
- Kuras, Z.; Yun, Y.H.; Chimote, A.A.; Neumeier, L.; Conforti, L. Kca3.1 and trpm7 channels at the uropod regulate migration of activated human t cells. PLoS ONE 2012, 7, e43859.
- Su, L.T.; Liu, W.; Chen, H.C.; Gonzalez-Pagan, O.; Habas, R.; Runnels, L.W. Trpm7 regulates polarized cell movements. Biochem. J. 2011, 434, 513–521.
- Chen, J.P.; Luan, Y.; You, C.X.; Chen, X.H.; Luo, R.C.; Li, R. Trpm7 regulates the migration of human nasopharyngeal carcinoma cell by mediating ca(2+) influx. Cell Calcium 2010, 47, 425–432.
- Desai, B.N.; Krapivinsky, G.; Navarro, B.; Krapivinsky, L.; Carter, B.C.; Febvay, S.; Delling, M.; Penumaka, A.; Ramsey, I.S.; Manasian, Y.; et al. Cleavage of trpm7 releases the kinase domain from the ion channel and regulates its participation in fas-induced apoptosis. Dev. Cell 2012, 22, 1149–1162.
- Chen, K.H.; Xu, X.H.; Liu, Y.; Hu, Y.; Jin, M.W.; Li, G.R. Trpm7 channels regulate proliferation and adipogenesis in 3t3-l1 preadipocytes. J. Cell Physiol. 2013, 229, 60–67.
- Zierler, S.; Yao, G.; Zhang, Z.; Kuo, W.C.; Porzgen, P.; Penner, R.; Horgen, F.D.; Fleig, A. Waixenicin a inhibits cell proliferation through magnesium-dependent block of transient receptor potential melastatin 7 (trpm7) channels. J. Biol. Chem. 2011, 286, 39328–39335.
- Zhang, Z.; Wang, M.; Fan, X.H.; Chen, J.H.; Guan, Y.Y.; Tang, Y.B. Upregulation of trpm7 channels by angiotensin ii triggers phenotypic switching of vascular smooth muscle cells of ascending aorta. Circ. Res. 2012, 111, 1137–1146.
- Abed, E.; Martineau, C.; Moreau, R. Role of melastatin transient receptor potential 7 channels in the osteoblastic differentiation of murine mc3t3 cells. Calcif. Tissue Int. 2011, 88, 246–253.
- Bernhardt, M.L.; Stein, P.; Carvacho, I.; Krapp, C.; Ardestani, G.; Mehregan, A.; Umbach, D.M.; Bartolomei, M.S.; Fissore, R.A.; Williams, C.J. Trpm7 and cav3.2 channels mediate ca(2+) influx required for egg activation at fertilization. Proc. Natl. Acad. Sci. USA 2018, 115, E10370–E10378.
- Carvacho, I.; Ardestani, G.; Lee, H.C.; McGarvey, K.; Fissore, R.A.; Lykke-Hartmann, K. Trpm7-like channels are functionally expressed in oocytes and modulate post-fertilization embryo development in mouse. Sci. Rep. 2016, 6, 34236.
- Michelle Aarts; Koji Iihara; Wen-Li Wei; Zhi-Gang Xiong; Mark Arundine; Waldy Cerwinski; John F. Macdonald; Michael Tymianski; A Key Role for TRPM7 Channels in Anoxic Neuronal Death. Cell 2003, 115, 863-877, 10.1016/s0092-8674(03)01017-1.
- Touyz, R.M. Transient receptor potential melastatin 6 and 7 channels, magnesium transport, and vascular biology: Implications in hypertension. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1103–H1118.
- Antunes, T.T.; Callera, G.E.; He, Y.; Yogi, A.; Ryazanov, A.G.; Ryazanova, L.V.; Zhai, A.; Stewart, D.J.; Shrier, A.; Touyz, R.M. Transient receptor potential melastatin 7 cation channel kinase: New player in angiotensin ii-induced hypertension. Hypertension 2016, 67, 763–773
- Hermosura, M.C.; Nayakanti, H.; Dorovkov, M.V.; Calderon, F.R.; Ryazanov, A.G.; Haymer, D.S.; Garruto, R.M. A trpm7 variant shows altered sensitivity to magnesium that may contribute to the pathogenesis of two guamanian neurodegenerative disorders. Proc. Natl. Acad. Sci. USA 2005, 102, 11510–11515.
- Tseveleki, V.; Rubio, R.; Vamvakas, S.S.; White, J.; Taoufik, E.; Petit, E.; Quackenbush, J.; Probert, L. Comparative gene expression analysis in mouse models for multiple sclerosis, alzheimer’s disease and stroke for identifying commonly regulated and disease-specific gene changes. Genomics 2010, 96, 82–91.
- Rios, F.J.; Zou, Z.G.; Harvey, A.P.; Harvey, K.Y.; Nosalski, R.; Anyfanti, P.; Camargo, L.L.; Lacchini, S.; Ryazanov, A.G.; Ryazanova, L.; et al. Chanzyme trpm7 protects against cardiovascular inflammation and fibrosis. Cardiovasc. Res. 2020, 116, 721–735.
- Sontia, B.; Montezano, A.C.; Paravicini, T.; Tabet, F.; Touyz, R.M. Downregulation of renal trpm7 and increased inflammation and fibrosis in aldosterone-infused mice: Effects of magnesium. Hypertension 2008, 51, 915–921.
- Suzuki, S.; Penner, R.; Fleig, A. Trpm7 contributes to progressive nephropathy. Sci. Rep. 2020, 10, 2333.
- Du, J.; Xie, J.; Zhang, Z.; Tsujikawa, H.; Fusco, D.; Silverman, D.; Liang, B.; Yue, L. Trpm7-mediated ca2+ signals confer fibrogenesis in human atrial fibrillation. Circ. Res. 2010, 106, 992–1003.
- Guilbert, A.; Gautier, M.; Dhennin-Duthille, I.; Haren, N.; Sevestre, H.; Ouadid-Ahidouch, H. Evidence that trpm7 is required for breast cancer cell proliferation. Am. J. Physiol. Cell Physiol. 2009, 297, C493–C502.
- Kim, B.J.; Park, E.J.; Lee, J.H.; Jeon, J.H.; Kim, S.J.; So, I. Suppression of transient receptor potential melastatin 7 channel induces cell death in gastric cancer. Cancer Sci. 2008, 99, 2502–2509.
- Jiang, J.; Li, M.H.; Inoue, K.; Chu, X.P.; Seeds, J.; Xiong, Z.G. Transient receptor potential melastatin 7-like current in human head and neck carcinoma cells: Role in cell proliferation. Cancer Res. 2007, 67, 10929–10938.
- Hanano, T.; Hara, Y.; Shi, J.; Morita, H.; Umebayashi, C.; Mori, E.; Sumimoto, H.; Ito, Y.; Mori, Y.; Inoue, R. Involvement of trpm7 in cell growth as a spontaneously activated ca2+ entry pathway in human retinoblastoma cells. J. Pharmacol. Sci. 2004, 95, 403–419.
- Middelbeek, J.; Kuipers, A.J.; Henneman, L.; Visser, D.; Eidhof, I.; van Horssen, R.; Wieringa, B.; Canisius, S.V.; Zwart, W.; Wessels, L.F.; et al. Trpm7 is required for breast tumor cell metastasis. Cancer Res. 2012, 72, 4250–4261.
- Rybarczyk, P.; Gautier, M.; Hague, F.; Dhennin-Duthille, I.; Chatelain, D.; Kerr-Conte, J.; Pattou, F.; Regimbeau, J.M.; Sevestre, H.; Ouadid-Ahidouch, H. Transient receptor potential melastatin-related 7 channel is overexpressed in human pancreatic ductal adenocarcinomas and regulates human pancreatic cancer cell migration. Int. J. Cancer 2012, 131, E851–E861.
- Chen, Y.F.; Chen, Y.T.; Chiu, W.T.; Shen, M.R. Remodeling of calcium signaling in tumor progression. J. Biomed. Sci. 2013, 20, 23.
- Gao, H.; Chen, X.; Du, X.; Guan, B.; Liu, Y.; Zhang, H. Egf enhances the migration of cancer cells by up-regulation of trpm7. Cell Calcium 2011, 50, 559–568
- Andrea Romagnani; Valentina Vettore; Tanja Rezzonico-Jost; Sarah Hampe; Elsa Rottoli; Wiebke Nadolni; Michela Perotti; Melanie A. Meier; Constanze Hermanns; Sheila Geiger; et al.Gunther WennemuthCamilla RecordatiMasayuki MatsushitaSusanne MuehlichMichele ProiettiVladimir ChubanovThomas GudermannFabio GrassiSusanna Zierler TRPM7 kinase activity is essential for T cell colonization and alloreactivity in the gut.. Nature Communications 2017, 8, 1917, 10.1038/s41467-017-01960-z.
- Simon Stritt; Paquita Nurden; Remi Favier; Marie Favier; Silvia Ferioli; Sanjeev K. Gotru; Judith M M. Van Eeuwijk; Harald Schulze; Alan T. Nurden; Michele P. Lambert; et al.Ernest TurroStephanie Burger-StrittMasayuki MatsushitaLorenz MittermeierPaola BalleriniSusanna ZierlerMichael A. LaffanVladimir ChubanovThomas GudermannBernhard NieswandtAndrew P. Braun Defects in TRPM7 channel function deregulate thrombopoiesis through altered cellular Mg2+ homeostasis and cytoskeletal architecture. Nature Communications 2016, 7, 11097, 10.1038/ncomms11097.
- Jin, J.; Desai, B.N.; Navarro, B.; Donovan, A.; Andrews, N.C.; Clapham, D.E. Deletion of trpm7 disrupts embryonic development and thymopoiesis without altering mg2+ homeostasis. Science 2008, 322, 756–760.
- Elizondo, M.R.; Arduini, B.L.; Paulsen, J.; MacDonald, E.L.; Sabel, J.L.; Henion, P.D.; Cornell, R.A.; Parichy, D.M. Defective skeletogenesis with kidney stone formation in dwarf zebrafish mutant for trpm7. Curr. Biol. 2005, 15, 667–671.
- Jin, J.; Wu, L.J.; Jun, J.; Cheng, X.; Xu, H.; Andrews, N.C.; Clapham, D.E. The channel kinase, trpm7, is required for early embryonic development. Proc. Natl. Acad. Sci. USA 2012, 109, E225–E233.
- Overton, J.D.; Komiya, Y.; Mezzacappa, C.; Nama, K.; Cai, N.; Lou, L.; Fedeles, S.V.; Habas, R.; Runnels, L.W. Hepatocystin is essential for trpm7 function during early embryogenesis. Sci. Rep. 2015, 5, 18395.
- Rajan Sah; Pietro Mesirca; Xenos Mason; William Gibson; Christopher Bates-Withers; Marjolein Van Den Boogert; Dipayan Chaudhuri; William T. Pu; Matteo E. Mangoni; David E. Clapham; et al. Timing of Myocardial Trpm7 Deletion During Cardiogenesis Variably Disrupts Adult Ventricular Function, Conduction, and Repolarization. Circulation 2013, 128, 101-114, 10.1161/circulationaha.112.000768.
- Lorenz Mittermeier; Lusine Demirkhanyan; Benjamin Stadlbauer; Andreas Breit; Camilla Recordati; Anne Hilgendorff; Masayuki Matsushita; Attila Braun; David G. Simmons; Eleonora Zakharian; et al.Thomas GudermannVladimir Chubanov TRPM7 is the central gatekeeper of intestinal mineral absorption essential for postnatal survival. Proceedings of the National Academy of Sciences 2019, 116, 4706-4715, 10.1073/pnas.1810633116.
- Susanna Zierler; Adriana Sumoza-Toledo; Sayuri Suzuki; Fionán Ó Dúill; Lillia V. Ryazanova; Reinhold Penner; Alexey G. Ryazanov; Andrea Fleig; TRPM7 kinase activity regulates murine mast cell degranulation. The Journal of Physiology 2016, 594, 2957-2970, 10.1113/jp271564.
- Katherine Brandao; Francina Deason-Towne; Xiaoyun Zhao; Anne-Laure Perraud; Carsten Schmitz; TRPM6 kinase activity regulates TRPM7 trafficking and inhibits cellular growth under hypomagnesic conditions. Cellular and Molecular Life Sciences 2014, 71, 4853-4867, 10.1007/s00018-014-1647-7.
- M. V. Dorovkov; A. G. Ryazanov; Phosphorylation of Annexin I by TRPM7 Channel-Kinase. Journal of Biological Chemistry 2004, 279, 50643-50646, 10.1074/jbc.c400441200.
- Kristopher Clark; Jeroen Middelbeek; Edwin Lasonder; Natalya G. Dulyaninova; Nick A. Morrice; Alexey G. Ryazanov; Anne R. Bresnick; Carl G. Figdor; Frank N. Van Leeuwen; TRPM7 Regulates Myosin IIA Filament Stability and Protein Localization by Heavy Chain Phosphorylation. Journal of Molecular Biology 2008, 378, 790-803, 10.1016/j.jmb.2008.02.057.
- Anne-Laure Perraud; Xiaoyun Zhao; Alexey G. Ryazanov; Carsten Schmitz; The channel-kinase TRPM7 regulates phosphorylation of the translational factor eEF2 via eEF2-k. Cellular Signalling 2011, 23, 586-593, 10.1016/j.cellsig.2010.11.011.
- M V Dorovkov; S N Beznosov; S Shah; L Kotlianskaia; A S Kostiukova; [Effect of mutations imitating the phosphorylation by TRPM7 kinase on the function of the N-terminal domain of tropomodulin].. Биофизика 2009, 53, 943–949.
- Francina Deason-Towne; Anne-Laure Perraud; Carsten Schmitz; Identification of Ser/Thr phosphorylation sites in the C2-domain of phospholipase C γ2 (PLCγ2) using TRPM7-kinase. Cellular Signalling 2012, 24, 2070-2075, 10.1016/j.cellsig.2012.06.015.
- Sandra Voringer; Laura Schreyer; Wiebke Nadolni; Melanie A. Meier; Katharina Woerther; Constanze Mittermeier; Silvia Ferioli; Stephan Singer; Kerstin Holzer; Susanna Zierler; et al.Vladimir ChubanovBernhard LieblThomas GudermannSusanne Muehlich Inhibition of TRPM7 blocks MRTF/SRF-dependent transcriptional and tumorigenic activity. Oncogene 2019, 39, 2328-2344, 10.1038/s41388-019-1140-8.
- Na Cai; Zhiyong Bai; Vikas Nanda; Loren W. Runnels; Mass Spectrometric Analysis of TRPM6 and TRPM7 Phosphorylation Reveals Regulatory Mechanisms of the Channel-Kinases. Scientific Reports 2017, 7, 42739, 10.1038/srep42739.
- Clark, K.; Middelbeek, J.; Morrice, N.A.; Figdor, C.G.; Lasonder, E.; van Leeuwen, F.N. Massive autophosphorylation of the ser/thr-rich domain controls protein kinase activity of trpm6 and trpm7. PLoS ONE 2008, 3, e1876.
- Matsushita, M.; Kozak, J.A.; Shimizu, Y.; McLachlin, D.T.; Yamaguchi, H.; Wei, F.Y.; Tomizawa, K.; Matsui, H.; Chait, B.T.; Cahalan, M.D.; et al. Channel function is dissociated from the intrinsic kinase activity and autophosphorylation of trpm7/chak1. J. Biol. Chem. 2005, 280, 20793–20803.
- Grigory Krapivinsky; Luba Krapivinsky; Yunona Manasian; David E. Clapham; The TRPM7 Chanzyme Is Cleaved to Release a Chromatin-Modifying Kinase. Cell 2014, 157, 1061-1072, 10.1016/j.cell.2014.03.046.
- Kaitsuka, T.; Katagiri, C.; Beesetty, P.; Nakamura, K.; Hourani, S.; Tomizawa, K.; Kozak, J.A.; Matsushita, M. Inactivation of trpm7 kinase activity does not impair its channel function in mice. Sci Rep. 2014, 4, 5718.
- Ryazanova, L.V.; Hu, Z.; Suzuki, S.; Chubanov, V.; Fleig, A.; Ryazanov, A.G. Elucidating the role of the trpm7 alpha-kinase: Trpm7 kinase inactivation leads to magnesium deprivation resistance phenotype in mice. Sci. Rep. 2014, 4, 7599.
- Byung Joo Kim; Ju-Hong Jeon; Seon Jeong Kim; InSuk So; Ki Whan Kim; Regulation of transient receptor potential melastatin 7 (TRPM7) currents by mitochondria.. Molecules and Cells 2007, 23, 363–369.
- Mahealani K. Monteilh-Zoller; Meredith C. Hermosura; Monica J.S. Nadler; Andrew M. Scharenberg; Reinhold Penner; Andrea Fleig; TRPM7 Provides an Ion Channel Mechanism for Cellular Entry of Trace Metal Ions. Journal of General Physiology 2002, 121, 49-60, 10.1085/jgp.20028740.
- Murali Prakriya; Richard S. Lewis; Separation and characterization of currents through store-operated CRAC channels and Mg2+-inhibited cation (MIC) channels.. Journal of General Physiology 2002, 119, 487–507.
- Xuanmao Chen; Tomohiro Numata; Minghua Li; Yasuo Mori; Beverley A. Orser; Michael F Jackson; Zhi-Gang Xiong; John F. Macdonald; The modulation of TRPM7 currents by nafamostat mesilate depends directly upon extracellular concentrations of divalent cations. Molecular Brain 2010, 3, 38-38, 10.1186/1756-6606-3-38.
- Parnas, M.; Peters, M.; Dadon, D.; Lev, S.; Vertkin, I.; Slutsky, I.; Minke, B. Carvacrol is a novel inhibitor of drosophila trpl and mammalian trpm7 channels. Cell Calcium 2009, 45, 300–309.
- Macianskiene, R.; Martisiene, I.; Zablockaite, D.; Gendviliene, V. Characterization of mg(2)(+)-regulated trpm7-like current in human atrial myocytes. J. Biomed. Sci 2012, 19, 75.
- Chen, W.L.; Barszczyk, A.; Turlova, E.; Deurloo, M.; Liu, B.; Yang, B.B.; Rutka, J.T.; Feng, Z.P.; Sun, H.S. Inhibition of trpm7 by carvacrol suppresses glioblastoma cell proliferation, migration and invasion. Oncotarget 2015, 6, 16321–16340.
- Chen, W.; Xu, B.; Xiao, A.; Liu, L.; Fang, X.; Liu, R.; Turlova, E.; Barszczyk, A.; Zhong, X.; Sun, C.L.; et al. Trpm7 inhibitor carvacrol protects brain from neonatal hypoxic-ischemic injury. Mol. Brain 2015, 8, 11.
- Luo, Y.; Wu, J.Y.; Lu, M.H.; Shi, Z.; Na, N.; Di, J.M. Carvacrol alleviates prostate cancer cell proliferation, migration, and invasion through regulation of pi3k/akt and mapk signaling pathways. Oxid Med. Cell Longev 2016, 2016, 1469693.
- Chen, H.C.; Xie, J.; Zhang, Z.; Su, L.T.; Yue, L.; Runnels, L.W. Blockade of trpm7 channel activity and cell death by inhibitors of 5-lipoxygenase. PLoS ONE 2010, 5, e11161.
- Kim, B.J.; Nam, J.H.; Kim, S.J. Effects of transient receptor potential channel blockers on pacemaker activity in interstitial cells of cajal from mouse small intestine. Mol. Cells 2011, 32, 153–160.
- Kim, B.J.; Kim, S.Y.; Lee, S.; Jeon, J.H.; Matsui, H.; Kwon, Y.K.; Kim, S.J.; So, I. The role of transient receptor potential channel blockers in human gastric cancer cell viability. Can. J. Physiol. Pharmacol. 2012, 90, 175–186.
- Mason, M.J.; Schaffner, C.; Floto, R.A.; Teo, Q.A. Constitutive expression of a mg2+-inhibited k+ current and a trpm7-like current in human erythroleukemia cells. Am. J. Physiol. Cell Physiol. 2012, 302, C853–C867
- Chen, J.; Dou, Y.; Zheng, X.; Leng, T.; Lu, X.; Ouyang, Y.; Sun, H.; Xing, F.; Mai, J.; Gu, J.; et al. Trpm7 channel inhibition mediates midazolam-induced proliferation loss in human malignant glioma. Tumour Biol. 2016, 37, 14721–14731.
- Dou, Y.; Li, Y.; Chen, J.; Wu, S.; Xiao, X.; Xie, S.; Tang, L.; Yan, M.; Wang, Y.; Lin, J.; et al. Inhibition of cancer cell proliferation by midazolam by targeting transient receptor potential melastatin 7. Oncol. Lett. 2013, 5, 1010–1016.
- Byung Joo Kim; Seung-Yeol Nah; Ju-Hong Jeon; InSuk So; Seon Jeong Kim; Transient Receptor Potential Melastatin 7 Channels are Involved in Ginsenoside Rg3-Induced Apoptosis in Gastric Cancer Cells. Basic & Clinical Pharmacology & Toxicology 2011, 109, 233-239, 10.1111/j.1742-7843.2011.00706.x.
- Kim, B.J. Involvement of melastatin type transient receptor potential 7 channels in ginsenoside rd-induced apoptosis in gastric and breast cancer cells. J. Ginseng Res. 2013, 37, 201–209.
- Zhang, Y.; Zhou, L.; Zhang, X.; Bai, J.; Shi, M.; Zhao, G. Ginsenoside-rd attenuates trpm7 and asic1a but promotes asic2a expression in rats after focal cerebral ischemia. Neurol. Sci. 2012, 33, 1125–1131.
- Mina Sato-Kasai; Takahiro A. Kato; Masahiro Ohgidani; Yoshito Mizoguchi; Noriaki Sagata; Shogo Inamine; Hideki Horikawa; Kohei Hayakawa; Norihiro Shimokawa; Sota Kyuragi; et al.Yoshihiro SekiAkira MonjiShigenobu Kanba Aripiprazole inhibits polyI:C-induced microglial activation possibly via TRPM7. Schizophrenia Research 2016, 178, 35-43, 10.1016/j.schres.2016.08.022.
- W. Nörenberg; Tanja Plötz; Helga Sobottka; Vladimir Chubanov; Lorenz Mittermeier; Hermann Kalwa; Achim Aigner; Michael Schaefer; TRPM7 is a molecular substrate of ATP-evoked P2X7-like currents in tumor cells. Journal of General Physiology 2016, 147, 467-483, 10.1085/jgp.201611595.
- Vladimir Chubanov; M Mederos Y Schnitzler; M Meißner; S Schäfer; Kathrin Abstiens; T Hofmann; T Gudermann; Natural and synthetic modulators of SK (Kca2) potassium channels inhibit magnesium-dependent activity of the kinase-coupled cation channel TRPM7. British Journal of Pharmacology 2012, 166, 1357-1376, 10.1111/j.1476-5381.2012.01855.x.
- Chubanov, V.; Ferioli, S.; Gudermann, T. Assessment of trpm7 functions by drug-like small molecules. Cell Calcium 2017, 67, 166–173.
- Chubanov, V.; Schafer, S.; Ferioli, S.; Gudermann, T. Natural and synthetic modulators of the trpm7 channel. Cells 2014, 3, 1089–1101
- Xin Qin; Zhichao Yue; Baonan Sun; Wenzhong Yang; Jia Xie; Eric Ni; Yi Feng; Rafat Mahmood; Yanhui Zhang; Lixia Yue; et al. Sphingosine and FTY720 are potent inhibitors of the transient receptor potential melastatin 7 (TRPM7) channels. British Journal of Pharmacology 2013, 168, 1294-1312, 10.1111/bph.12012.
- T. Hofmann; S. Schafer; M. Linseisen; L. Sytik; T. Gudermann; Vladimir Chubanov; Activation of TRPM7 channels by small molecules under physiological conditions. Pflügers Archiv - European Journal of Physiology 2014, 466, 2177-2189, 10.1007/s00424-014-1488-0.
- Sebastian Schäfer; Silvia Ferioli; Thomas Hofmann; Susanna Zierler; Thomas Gudermann; Vladimir Chubanov; Mibefradil represents a new class of benzimidazole TRPM7 channel agonists. Pflügers Archiv - European Journal of Physiology 2015, 468, 623-634, 10.1007/s00424-015-1772-7.
- John Doukas; Wolfgang Wrasidlo; Glenn Noronha; Elena Dneprovskaia; Richard Fine; Sara Weis; John Hood; Anthony DeMaria; Richard Soll; David Cheresh; et al. Phosphoinositide 3-kinase / inhibition limits infarct size after myocardial ischemia/reperfusion injury. Proceedings of the National Academy of Sciences 2006, 103, 19866-19871, 10.1073/pnas.0606956103.
- Mindy I Davis; Jeremy P Hunt; Sanna Herrgard; Pietro Ciceri; Lisa M Wodicka; Gabriel Pallares; Michael Hocker; Daniel K Treiber; Patrick P Zarrinkar; Comprehensive analysis of kinase inhibitor selectivity. Nature Biotechnology 2011, 29, 1046-1051, 10.1038/nbt.1990.
- Chiman Song; Yeonju Bae; Jinjoo Jun; Hyomin Lee; Nam Doo Kim; Kyung-Bok Lee; Wooyoung Hur; Jae-Yong Park; Taebo Sim; Identification of TG100-115 as a new and potent TRPM7 kinase inhibitor, which suppresses breast cancer cell migration and invasion. Biochimica et Biophysica Acta (BBA) - General Subjects 2017, 1861, 947-957, 10.1016/j.bbagen.2017.01.034.

