 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Silvia Gazzin | + 965 word(s) | 2859 | 2020-09-29 08:24:26 | | | |
| 2 | Camila Xu | Meta information modification | 965 | 2020-10-15 11:02:25 | | | | |
| 3 | Camila Xu | + 3 word(s) | 968 | 2020-10-21 11:06:44 | | | | |
| 4 | Camila Xu | + 3 word(s) | 968 | 2020-10-21 11:07:06 | | | | |
| 5 | Camila Xu | + 3 word(s) | 968 | 2020-10-21 11:07:29 | | | | |
| 6 | Catherine Yang | Meta information modification | 968 | 2021-09-28 08:41:50 | | |
Video Upload Options
Bilirubin is a yellow endogenous derivate of the heme catabolism. Since the 1980s, it has been recognized as one of the most potent antioxidants in nature, able to counteract 10,000× higher intracellular concentrations of H2O2. In the recent years, not only bilirubin, but also its precursor biliverdin, and the enzymes involved in their productions (namely heme oxygenase and biliverdin reductase; altogether the "yellow players"-YPs) have been recognized playing a protective role in diseases characterized by a chronic prooxidant status.
1. Introduction
Bilirubin, the end product of the consecutive enzymatic activity of heme oxygenase (HMOX) and biliverdin reductase (BLVR), is mostly known as a serum marker of hepatic diseases [1][2]. Bilirubin circulates in the blood in its unconjugated form (UCB, unconjugated bilirubin) tightly bound to albumin, with a minimal portion being unbound (free bilirubin, Bf, about 0.1% in physiological conditions) [3], and is mainly produced from heme, originating from the senescent red blood cells in the spleen. UCB is highly hydrophobic and potentially toxic in high concentrations [4][5][6], and is conjugated in the liver with 1 or 2 molecules of glucuronic acid. The formed polar conjugated bilirubin (CB), after its further metabolism in the gut lumen, is easily discarded from the body though feces. Defects in hepatic conjugation will increase the UCB content in blood, with consequent rise of the Bf fraction in serum when UCB concentration exceed the capacity of its binding compounds [3].
2. Specifics
Due to its lipophilic properties, Bf may diffuse across the cellular bilayer entering the cells. Based on this classic concept, the blood supply has been for a longtime considered the unique source of bilirubin content in the extrahepatic tissues, including the central nervous system (CNS) (Figure 1) [7][8].
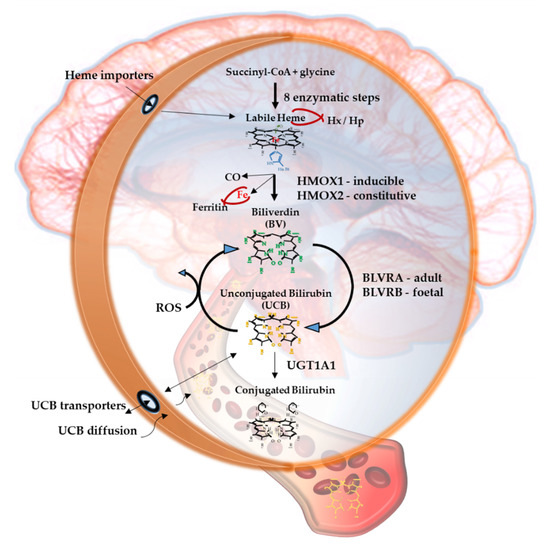
Figure 1. The Yellow Players.*YPs
When entering cells, UCB may counteract 10,000× higher concentrations of H2O2, being one of the most potent antioxidants in nature [3][9]. For a long time the explanation of this incredible antioxidant ability has been based on the concept of the bilirubin-biliverdin redox cycle (Figure 1), where bilirubin is oxidized back to its precursor biliverdin (BV) by reactive oxygen species (ROS), and, in turn, BV is rapidly reduced by BLVR to bilirubin [10]. As a result, the antioxidant effects of UCB is amplified without increasing the cellular concentration of the pigment to a toxic level.
Figure 1 resumes the main steps of bilirubin metabolism, as well as the basis for its antioxidant capability. The concentration of systemic (blood) bilirubin derives from the transformation of the intracellular heme (the so-called labile heme) into biliverdin (BV), together with CO and Fe2+, by the action of heme oxygenase (HMOX) enzymes. BV is then converted into unconjugated bilirubin (UCB) by the enzyme biliverdin reductase (BLVR). Transported to the liver by blood, UCB hydrophobic and toxic in high concentrations, is then conjugated by the uridine diphospho-glucuronosyl transferase (UGT) 1A1 to conjugated bilirubin (CB), and eliminated from the body. Inside the cell, the powerful antioxidant action of UCB is due to its conversion back to BV during the scavenging of the cellular ROS. In this BV-bilirubin redox cycle, the protection is continuously renewed maintaining the intracellular physiological concentration of the pigments. Based on this traditional concept, the main source of labile heme (thus UCB) is the turnover of the senescent red blood cells in the spleen, and the intracellular concentration of UCB in extrahepatic tissues is believed to depend on blood supply. If true, it may account for even toxic supply of heme and UCB in case of stroke or CNS conditions compromising the blood-brain interfaces. Nevertheless, recent data suggest that extrahepatic cells may produce de novo UCB, starting from a pool of labile heme that might also be replenished from both an import, as well as an in situ (intracellular) synthesis. Added to the ubiquitarian on-demand induction of HMOX and BLVR under stressor stimuli, the YPs form a local homeostatic and defensive cellular system, that might act in synergy or independently from the systemic blood bilirubin, with hemopexin (Hx), haptoglobin (Hp), and ferritin preventing the generation of ROS by the chelating/binding of free hemoglobin and iron.
Based on the recent experimental as well as clinical data not only of UCB but also of the enzymes and precursors involved in its production seem to be importantly implemented in the pathogenesis of CNS’s disorders.
Both HMOX and BLVR possess multiple binding sites for transcription factors on the promoter region of the gene, making them able to react on demand to stressor stimuli, including those characterizing the diseases [11][12][13][14][15][16], pointing to an active role in the cellular defense. In line with this concept is their induction described in several pathological conditions [1][17].
3. Current status
Recently, different cell types (including neuronal cells), have been demonstrated in vitro to be able to produce de novo bilirubin from its precursors, increasing cellular resistance to damage [18][19][20]. In eels, UCB cellular production and storage (UCB bind to a protein named UnaG, belonging to the fatty acid-binding protein (FABP) family) have been suggested to provide a cellular homeostatic system able to face the oxidative challenge of the eel migration [21][22]. This has not only confirmed the idea of an active role of UCB in response to stress but has underlined the importance of the cellular UCB concentration in this process.
Finally, a correlation between UCB concentration, as well as HMOX1/BLVR activation, and the diseases have been described both in the experimental and clinical studies [1][17].
Considering quite a specific environment of the CNS-highly lipophilic, with high O2 consumption and a limited expression of antioxidant defense, making the brain highly susceptible to oxidative stress—the modulation of bilirubin and the YPs may be an intriguing therapeutic target.
The vast majority of our current knowledge on the role of the YPs derives from extra CNS diseases (such as cardiovascular diseases, metabolic syndrome, diabetes, etc.), while what this entails specifically for the CNS is still largely unknown.
References
- Gazzin, S.; Vitek, L.; Watchko, J.; Shapiro, S.M.; Tiribelli, C. A Novel Perspective on the Biology of Bilirubin in Health and Disease. Trends Mol. Med. 2016, 22, 758–768. [Google Scholar] [CrossRef] [PubMed]
- Gazzin, S.; Masutti, F.; Vítek, L.; Tiribelli, C. The molecular basis of jaundice: An old symptom revisited. Liver Int. 2016, 37, 1094–1102. [Google Scholar] [CrossRef] [PubMed]
- Vítek, L.; Ostrow, J.D. Bilirubin Chemistry and Metabolism; Harmful and Protective Aspects. Available online: https://www.eurekaselect.com/69920/article (accessed on 27 July 2020).
- Le Pichon, J.-B.; Riordan, S.M.; Watchko, J.; Shapiro, S.M. The Neurological Sequelae of Neonatal Hyperbilirubinemia: Definitions, Diagnosis and Treatment of the Kernicterus Spectrum Disorders (KSDs). Curr. Pediatr. Rev. 2017, 13, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Strauss, K.A.; Robinson, D.L.; Vreman, H.J.; Puffenberger, E.G.; Hart, G.; Morton, D.H. Management of hyperbilirubinemia and prevention of kernicterus in 20 patients with Crigler-Najjar disease. Eur. J. Pediatr. 2006, 165, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Watchko, J.F.; Tiribelli, C. Bilirubin-Induced Neurologic Damage—Mechanisms and Management Approaches. N. Engl. J. Med. 2013, 369, 2021–2030. [Google Scholar] [CrossRef] [PubMed]
- Diamond, I.D.; Schmid, R.S. Experimental bilirubin encephalopathy. The mode of entry of bilirubin-14C into the central nervous system. J. Clin. Investig. 1966, 45, 678–689. [Google Scholar] [CrossRef]
- Wennberg, R.P.; Ahlfors, C.E.; Bhutani, V.K.; Johnson, L.H.; Shapiro, S.M. Toward Understanding Kernicterus: A Challenge to Improve the Management of Jaundiced Newborns. Pediatrics 2006, 117, 474–485. [Google Scholar] [CrossRef]
- Stocker, R.; Yamamoto, Y.; McDonagh, A.F.; Glazer, A.N.; Ames, B.N. Bilirubin is an antioxidant of possible physiological importance. Science 1987, 235, 1043–1046. [Google Scholar] [CrossRef]
- Baranano, D.E.; Rao, M.; Ferris, C.D.; Snyder, S.H. Biliverdin reductase: A major physiologic cytoprotectant. Proc. Natl. Acad. Sci. USA 2002, 99, 16093–16098. [Google Scholar] [CrossRef]
- Abraham, N.G.; Kappas, A. Pharmacological and Clinical Aspects of Heme Oxygenase. Pharmacol. Rev. 2008, 60, 79–127. [Google Scholar] [CrossRef]
- Gozzelino, R. The Pathophysiology of Heme in the Brain. Available online: https://www.eurekaselect.com/135089/article (accessed on 27 July 2020).
- Maines, M.D. New Insights into Biliverdin Reductase Functions: Linking Heme Metabolism to Cell Signaling. Physiology 2005, 20, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Nitti, M.; Piras, S.; Brondolo, L.; Marinari, U.M.; Pronzato, M.A.; Furfaro, A.L. Heme Oxygenase 1 in the Nervous System: Does It Favor Neuronal Cell Survival or Induce Neurodegeneration? Int. J. Mol. Sci. 2018, 19, 2260. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Alam, J.; Choi, A.M.K. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M.; Song, W.; Tavitian, A.; Cressatti, M. The sinister face of heme oxygenase-1 in brain aging and disease. Prog. Neurobiol. 2019, 172, 40–70. [Google Scholar] [CrossRef]
- Wagner, K.-H.; Wallner, M.; Mölzer, C.; Gazzin, S.; Bulmer, A.C.; Tiribelli, C.; Vitek, L. Looking to the horizon: The role of bilirubin in the development and prevention of age-related chronic diseases. Clin. Sci. 2015, 129, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Tu, Y.; Moon, C.; Nagata, E.; Ronnett, G.V. Heme oxygenase-1 and heme oxygenase-2 have distinct roles in the proliferation and survival of olfactory receptor neurons mediated by cGMP and bilirubin, respectively. J. Neurochem. 2003, 85, 1247–1261. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-S.; Nam, E.; Lee, H.-K.; Lim, M.H.; Rhee, H.-W. In Cellulo Mapping of Subcellular Localized Bilirubin. ACS Chem. Biol. 2016, 11, 2177–2185. [Google Scholar] [CrossRef]
- Takeda, T.; Mu, A.; Tai, T.T.; Kitajima, S.; Taketani, S. Continuous de novo biosynthesis of haem and its rapid turnover to bilirubin are necessary for cytoprotection against cell damage. Sci. Rep. 2015, 5, 10488. [Google Scholar] [CrossRef]
- Funahashi, A.; Komatsu, M.; Furukawa, T.; Yoshizono, Y.; Yoshizono, H.; Orikawa, Y.; Takumi, S.; Shiozaki, K.; Hayashi, S.; Kaminishi, Y.; et al. Eel green fluorescent protein is associated with resistance to oxidative stress. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2016, 181–182, 35–39. [Google Scholar] [CrossRef]
- Kumagai, A.; Ando, R.; Miyatake, H.; Greimel, P.; Kobayashi, T.; Hirabayashi, Y.; Shimogori, T.; Miyawaki, A. A Bilirubin-Inducible Fluorescent Protein from Eel Muscle. Cell 2013, 153, 1602–1611.

