Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rameez Hassan Pirzada | -- | 2485 | 2022-05-25 06:33:00 | | | |
| 2 | Catherine Yang | + 1 word(s) | 2486 | 2022-05-25 07:40:51 | | | | |
| 3 | Rameez Hassan Pirzada | Meta information modification | 2486 | 2022-05-25 12:21:22 | | | | |
| 4 | Sangdun Choi | Meta information modification | 2486 | 2022-05-25 13:06:19 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Pirzada, R.H.; Choi, S.; Javaid, N. NLRP3 Inflammasome in Neurodegenerative. Encyclopedia. Available online: https://encyclopedia.pub/entry/23327 (accessed on 01 August 2026).
Pirzada RH, Choi S, Javaid N. NLRP3 Inflammasome in Neurodegenerative. Encyclopedia. Available at: https://encyclopedia.pub/entry/23327. Accessed August 01, 2026.
Pirzada, Rameez Hassan, Sangdun Choi, Nasir Javaid. "NLRP3 Inflammasome in Neurodegenerative" Encyclopedia, https://encyclopedia.pub/entry/23327 (accessed August 01, 2026).
Pirzada, R.H., Choi, S., & Javaid, N. (2022, May 25). NLRP3 Inflammasome in Neurodegenerative. In Encyclopedia. https://encyclopedia.pub/entry/23327
Pirzada, Rameez Hassan, et al. "NLRP3 Inflammasome in Neurodegenerative." Encyclopedia. Web. 25 May, 2022.
Copy Citation
Inflammasomes are intracellular multiprotein complexes in the cytoplasm that regulate inflammation activation in the innate immune system in response to pathogens and to host self-derived molecules. The NLRP3 belongs to the subfamily of NLRP which activates caspase 1, thus causing the production of proinflammatory cytokines (interleukin 1β and interleukin 18) and pyroptosis. This inflammasome is involved in multiple neurodegenerative and metabolic disorders including Alzheimer’s disease, multiple sclerosis, type 2 diabetes mellitus, and gout.
inflammasome
Alzheimer’s disease
type 2 diabetes mellitus
1. The NLRP3 Inflammasome
NLRP3 was initially characterized in an autoinflammatory disease named Muckle–Wells syndrome [1]. The NLRP3 inflammasome complex is mainly composed of three units: a receptor protein (NLRP3), an adaptor protein (ASC), and an effector protein (caspase 1) [2][3]. The receptor protein acts as a sensor that is switched on after sensing a PAMP and/or DAMP. The ASC adaptor protein contains two death domains: The N-terminal pyrin domain (PYD) and the C-terminal caspase recruitment domain (CARD) [3][4], which serves as a mediator between the sensor and effector protein.
The NLRP3 complex is primarily expressed in immune cells such as inflammatory and antigen-presenting cells (APCs). It is comprised of central nucleotide-binding and oligomerization domain (NBD), a C-terminal leucine-rich repeat LRR domain, and an N-terminal pyrin domain. NLRP3 as a receptor protein is stimulated by the presence of PAMPs and/or DAMPs including nucleic acids, lipooligosaccharide (LPS) and muramyl dipeptide (MDP) [5][6]. NLRP3 activation is divided into two steps: priming and activation [7]. The priming signal (first signal) includes a large number of DAMPs and/or PAMPs which activate PRR (such as TLR) signaling to stimulate the nuclear factor-κB (NFκB) pathway. This pathway promotes the transcription and expression of NLRP3 along with pro-IL-1β and pro-IL-18 that are translocated from the nucleus to cytoplasm in an inactive form [8]. Moreover, the priming signal includes injury-related factors such as oxidized low-density lipoprotein [9], whose accumulation has been identified in metabolically induced obesity-related diseases. The activating signal (second signal)—that initiates NLRP3 inflammasome stimulation—originates from a variety of activators such as PAMPs, DAMPs, exogenous adenosine, amyloid β (Aβ), mitochondrial DNA, or substances (e.g., asbestos and aluminum or silica and uric acid crystals) [10][11][12]. Furthermore, ATP-induced P2X7R activation raises K+ efflux, which is also critical for NLRP3 activation [13]. Eventually, ASC via the CARD domain mediates the recruitment of pro-caspase 1, which drives NLRP3 inflammasome assembly. The proximity to neighboring pro-caspase 1 leads to its autocleavage and conversion into mature caspase 1, which subsequently cleaves the precursor cytokines (pro-IL-1β and pro-IL-18) into their mature forms (IL-1β and IL-18) [14][15]. This event next initiates pyroptosis, an inflammatory type of programmed cell death that is controlled by gasdermin D (GSDMD) [16]. In pyroptosis, pores are formed in the plasma membrane as the N-terminal fragment of GSDMD detaches from its C-terminal inhibitory domain and attaches to phosphoinositides, which oligomerize to produce membrane pores [17][18]. The formation of membrane pores disturbs the cellular osmotic potential, thereby leading to cell swelling and lysis with the ultimate release of mature IL-1β and IL-18 into the extracellular environment [16].
So far, numerous factors have been found to initiate NLRP3 inflammasome activation; however, the exact mechanism remains unclear and represents an area of active investigation. A possible mechanism underlying the initiation of the NLRP3 inflammasome includes reactive oxygen species (ROS) production and mitochondrial dysfunction, oxidized mitochondrial DNA release, K+ efflux, a cathepsin B release from disrupted lysosomes, changes in extracellular Ca2+ ion gradients, and the formation of transmembrane pores [19][20][21][22][23][24]. Moreover, NIMA-related kinase 7 (NEK7) has been reported to bind to the LRR domain of NLRP3 thus leading to its activation and oligomerization (Figure 1) [25]. On the other hand, opposing evidence highlights the controversial role of mitochondrial ROS in the inhibition of NLRP3 activation [26]. Moreover, studies have revealed that post-translational modifications are significantly involved in NLRP3 activation [14][27][28]. Nonetheless, ubiquitination and post-translational modifications at the priming step have been found to induce NLRP3 inflammasome inactivation [29][30], whereas dephosphorylation and deubiquitination cause its activation [28][31]. Additionally, protein kinase A–associated NLRP3 phosphorylation on residue Ser291 is important for NLRP3 inflammasome inactivation [32]. Moreover, recent studies uncovered the function of microRNAs in the regulation of the NLRP3 inflammasome. For example, myeloid-derived miR-223 controls intestinal inflammation by repressing the NLRP3 inflammasome [33]. miR-33 has also been demonstrated to regulate the NLRP3 inflammasome in macrophages because this pathway plays a critical part in the development of rheumatoid arthritis [34]. This observation provides further insights into the events that epigenetic regulators induce in this inflammasome.
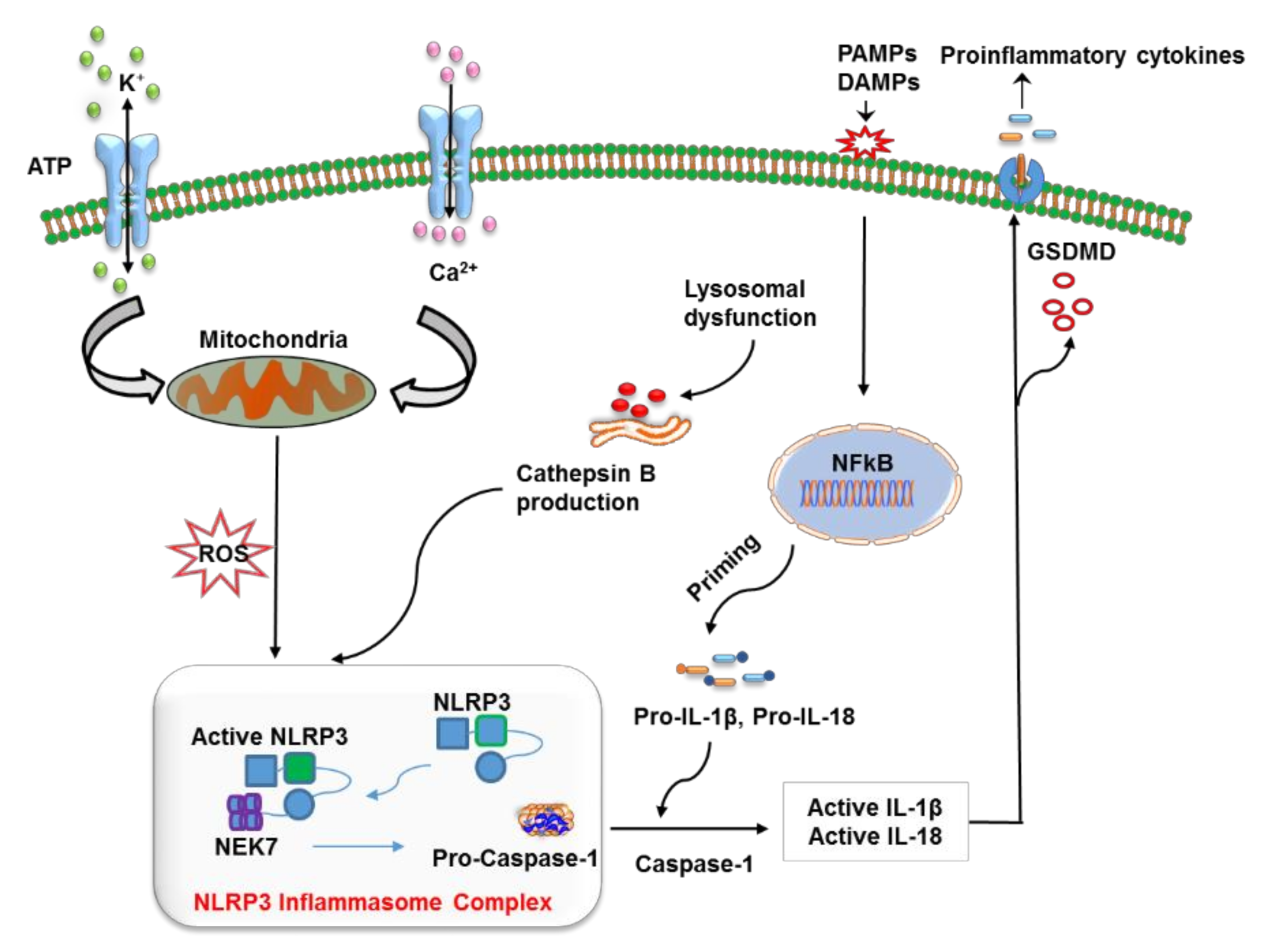
Figure 1. The mechanism of action of the nucleotide-binding oligomerization domain-like receptor (NLR) family pyrin domain containing 3 (NLRP3) inflammasome. The priming step involves the recognition of a pathogen-associated molecular pattern (PAMP) or a damage-associated molecular pattern (DAMP) by a specific pattern recognition receptors (PRR), which activates the NF-κB pathway to release precursor forms of IL-1β and IL-18 into the cytoplasm. NLRP3 is turned on by lysosome-mediated cathepsin B, K+ efflux, reactive oxygen species (ROS) production via dysfunctional mitochondria, the release of mitochondrial DNA in oxidized form, and alterations in Ca2+ concentration. The oligomerization and activation of NLRP3 take place after it interacts with the leucine-rich repeat (LRR) domain of NEK7. This event is followed by the cleavage of pro-caspase 1 into caspase 1, which converts pro-IL-1β and pro-IL-18 into their respective mature forms, which are finally released from the cell via pores generated by gasdermin D (GSDMD) (N-terminal fragments).
Thus, numerous stimuli are recognized by their receptors and serve as first, second, or both signals to make NLRP3 active; nevertheless, further research is necessary to determine the exact mechanism.
2. Roles of NLRP3 Inflammasomes in Metabolic and Neurodegenerative Diseases
NLRP3 was initially characterized as a major causative factor of inflammation owing to NLRP3’ involvement in a group of rare heterogeneous autoinflammatory conditions, known as cryopyrin-associated periodic syndrome [35]. On the other hand, its activation by damage-associated stimuli activates innate response to tissue damage. Therefore, inflammasomes significantly contribute to the pathogenesis of various diseases including dementia, multiple sclerosis, cancer, and gout [36][37][38]. The most common form of dementia (60–80%) is Alzheimer’s disease (AD) which leads to neurodegeneration by various mechanisms such as mitochondrial dysfunction, oxidative stress, and inflammation. Metabolic disorders such as obesity and type 2 diabetes (T2D) have a strong correlation to increased risk of AD [39][40][41]. This association involves increased accumulation of amyloid-β (Aβ) which is confirmed in various mouse models [42][43][44]. Obesity, T2D, and AD are affected by the involvement of NLRP3; here, the researchers focus on their association with each other from a therapeutic point of view.
2.1. Obesity and T2D
Anomalous activation of the innate immune system significantly contributes to the pathogenesis of metabolic disorders such as T2D [45][46]. The latter is a chronic condition that was formerly termed as adult-onset diabetes, which is characterized by hyperglycemia and—according to more recent insights—by relative insulin deficiency triggered by pancreatic β-cell dysfunction [47]. In addition to other factors, obesity is a major factor responsible for T2D worldwide. Its prevalence may further increase globally, with the highest projected prevalence rates in developing or low-income countries [48]. Therefore, deeper insights into obesity pathogenesis as a significant risk factor for T2D hold huge promise for obesity prevention and treatment [49][50].
Obesity has multifactorial pathogenesis, which includes the growth of adipocytes and increased infiltration of macrophages into adipose tissue (AT) thereby activating inflammatory pathways and causing chronic inflammation [51]. Obesity-induced alterations in adipocytes and macrophages cause insulin resistance (IR) with subsequent induction of AT fibrosis [52]. Besides energy conservation, ATs secrete adipokines, molecules that contribute (via endocrine, autocrine, and paracrine signaling mechanisms) to various physiological and pathophysiological conditions, thereby regulating inflammation and immunity, insulin sensitivity, and food intake [53]. Furthermore, a reduction in the secretion of insulin-sensitive adipokines accompanied by oversecretion of proinflammatory cytokines (TNF-α, IL-1β, and IL-6) is closely related to the etiology of various metabolic conditions. Other studies also point to the participation of NLRP3 in obesity because of its overexpression in AT [54][55][56][57][58][59]. Moreover, the results of studies on mouse models of obesity are consistent with this observation [60][61]. The release of proinflammatory cytokines (IL-1β and IL-18) is the main culprit behind the AT inflammation in obese subjects [62] and thus this contributes to the development of T2D. The detailed function of the NLRP3 inflammasome in the pathogenesis of T2D is explained in a systematic review [63].
The activation of innate-immunity cells (such as neutrophils and macrophages) owing to the emergence of a chronic proinflammatory state has been closely associated with IR [64][65]. Furthermore, the activation of inflammasomes is due to the priming signals produced by the engagement of PRRs such as TLRs [46][66][67]. These events contribute to the development of IR and liver fat storage in mice with diet-induced obesity because of macrophage infiltration into AT [68][69][70]. Additionally, overexpression of NLRP3 inflammasome components in AT is associated with the pathogenesis of obesity and therefore is directly associated with T2D, atherosclerosis, and myocardial infarction [54][71]. Increased amyloid polypeptide deposition in pancreatic islets is one of the key inducers of NLRP3 activation [72][73] via lysosomal disruption and high ROS concentrations in pancreas-infiltrating macrophages [74]. Additionally, in comparison to wild-type mice, NLRP3 knockout mice show substantial improvement in insulin sensitivity during a high-fat diet; this approach provides protection against obesity [54][75][76]. Activation of NLRP3 causes oversecretion of inflammatory cytokines such as IL-1β, which impair islet cell function and induce dysregulation of blood glucose levels, thus resulting in the development of T2D [77][78]. The surface expression of functional IL-1β receptors on pancreatic β cells and infiltrating macrophages further enhances the production of IL-1β and the diffusion of inflammatory signals through the NF-kB pathway, which might ultimately cause β-cell dysfunction [77][79]. Even the early stage of T2D is characterized by dysfunctional β cells and a decrease in insulin production.
2.2. Alzheimer’s Disease
AD is the most common neurodegenerative disorder. It is clinically characterized by dementia and progressive cognitive impairment, which are caused by two main pathologies: Aβ plaques and neurofibrillary tangles [80]. The estimated yearly incidence and prevalence of AD rise significantly with age. The incidence rate varies among different age groups: among people aged between 65 and 69, the approximate incidence is 0.4%; in people aged over 90, this rate is nearly 10% [81]. A significant contributing factor to the pathogenesis of AD includes aggregation of Aβ into neurotoxic plaques [82]. Accumulation of Aβ launches several cellular responses mediated by microglia, a type of neuroimmune cell located throughout the brain [83]. Recent studies revealed that Aβ can initiate NLRP3 inflammasome signaling in microglia [84], which causes the production of proinflammatory cytokines and consequently induces inflammation [85]. Studies have uncovered the involvement of several inflammatory components including prostaglandins, chemokines, and cytokines that are expressed in postmortem brain tissues isolated from patients with neurodegenerative disorders including AD; moreover, their elevated expression has been identified in cerebrospinal fluid [86][87]. Astrocytes and microglia are the mediators of innate immunity in the brain and they sense pathogens or other inflammatory signals; this property contributes to the activation and assembly of inflammasomes and ultimately leads to caspase-1–induced maturation of the IL-1β cytokine [88][89]. Although a normal level of IL-1β is present in a healthy brain, its overproduction may cause inflammation and the associated pathological complications.
On the other hand, various studies have revealed the overexpression of IL-1β in microglia in the vicinity of Aβ plaques in animal models and AD patients [90][91]. The microglial cells in the CNS phagocytose Aβ plaques; this process initiates lysosomal destabilization and the cytosolic production of cathepsin B, which may act as an endogenous signal and make NLRP3 active [84]. Aβ-mediated NLRP3 activation has been found to upregulate IL-1β, which promotes the formation of microglial cells and the accumulation of neurotoxic inflammatory factors [84][92]. Moreover, NLRP3 triggering plays a substantial role in the pathogenesis of amyloidosis. Similarly, in a transgenic mouse model of AD, an NLRP3 knockout protects from spatial memory dysfunction and reduces the deposition of Aβ [93]. The recruitment of microglia in AD is due to the secretion of neurotoxic components produced by senile neurofibrillary plaques [94]; these cells then phagocytose Aβ deposits and further secrete proinflammatory and chemotactic cytokines to aggravate the neurotoxic effects of Aβ. This observation further supports the functions performed by the activation of endogenous NLRP3 in the brain microglial cells because of formation of a plaque [93]. Therefore, the AD pathogenesis is strongly associated with the microglia-mediated NLRP3 inflammasome activation. Additionally, the restoration of Myb1 (TOM1) also reduces Aβ pathology, thereby highlighting the importance of endosomal adaptors and their associated factors in AD pathogenesis [95]. Depending on AD progression and stage, microglia upon activation adopt the M1 phenotype, and the overexpression of proinflammatory cytokines, such as IL-1β, induces an aberration in microglial cells; this event has a negative impact on the Aβ clearance system [96][97]. Nevertheless, an M2-like phenotype is associated with the production of anti-inflammatory cytokines, which contribute to protection against damage induced by inflammation and promote tissue remodeling [98]. Furthermore, activation of NLRP3 inflammasomes induces the extracellular release of ASC particles that act as DAMPs and consequently attract surrounding macrophages [99]. These micrometer-sized ASC specks have been reported to establish a bond with Aβ and promote plaque formation and misfolded protein accumulation, as observed in the APP/PS1 model of AD [100].
Moreover, the most reliable factor underlying the pathogenesis of AD is chronic localized neuroinflammation. Dysregulation of the interaction pattern between microglia and brain neurons is thought to be a contributing factor of AD pathogenesis [101][102]. The overproduction of cytotoxic molecules and proinflammatory cytokines by brain-resident immune cells, such as microglia, launches signaling pathways in neurons, consequently inducing brain tissue damage [103]. Numerous studies have pointed out that overexpression of IL-1β aggravates AD pathogenesis, owing to tau hyperphosphorylation [104], which inhibits long-term potentiation and affects synaptic plasticity [105][106]. Furthermore, IL-1β inhibition in a mouse model has yielded disease-modifying improvements in the pathology of AD [107]. A recent study suggests that a small-molecule NLRP3 inhibitor (JC-124) exerts a beneficial effect on a mouse model of AD [108], similarly to a knockout mouse model that manifested an improvement in spatial memory [93]. Neuronal damage results in the production of proinflammatory cytokines (IL-1β, TNF-α, and IL-6) and triggers apoptotic mitogen-activated protein kinase p38 (MAPK) [109]. P38 MAPK causes glutamate-mediated neurotoxicity via the N-methyl D-aspartic acid receptor [110][111]. Furthermore, IL-1β production induces nitric oxide synthase (iNOS) surrounding the hippocampus; this induction consequently causes damage and neuronal death [112]. IL-1β expression is significantly associated with NLRP3 inflammasome activation, and pyroptosis induced by the inflammasome is an active area of research. Accordingly, deep insight into the mechanism of NLRP3 inflammasome regulation is urgently needed to further unravel AD pathogenesis.
The aforementioned studies have provided significant insights into the role NLRP3 inflammasomes play in AD progression; therefore, detailed investigation into the mechanism of inflammasome activation and its contribution to neuroinflammation will be worthwhile. Moreover, Aβ accumulation induces microglial activation via advanced glycation end product (AGE) pathways and TLR [113][114]. All these results support the usefulness of NLRP3 inflammasomes and of their downstream regulators in the identification of protein aggregates or peptides that can drive the pathogenesis of such diseases as systemic amyloidosis, prion diseases, AD, and T2D. Targeting of components of the NLRP3 inflammasome, especially at the initial stages of the disease, may reduce the formation and aggregation of Aβ plaques to delay neurological damage in these patients.
References
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426.
- von Moltke, J.; Ayres, J.S.; Kofoed, E.M.; Chavarria-Smith, J.; Vance, R.E. Recognition of bacteria by inflammasomes. Annu. Rev. Immunol. 2013, 31, 73–106.
- Rathinam, V.A.; Fitzgerald, K.A. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell 2016, 165, 792–800.
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420.
- Kanneganti, T.D.; Body-Malapel, M.; Amer, A.; Park, J.H.; Whitfield, J.; Franchi, L.; Taraporewala, Z.F.; Miller, D.; Patton, J.T.; Inohara, N.; et al. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J. Biol. Chem. 2006, 281, 36560–36568.
- Marina-Garcia, N.; Franchi, L.; Kim, Y.G.; Miller, D.; McDonald, C.; Boons, G.J.; Nunez, G. Pannexin-1-mediated intracellular delivery of muramyl dipeptide induces caspase-1 activation via cryopyrin/NLRP3 independently of Nod2. J. Immunol. 2008, 180, 4050–4057.
- Ozaki, E.; Campbell, M.; Doyle, S.L. Targeting the NLRP3 inflammasome in chronic inflammatory diseases: Current perspectives. J. Inflamm. Res. 2015, 8, 15–27.
- Franchi, L.; Eigenbrod, T.; Munoz-Planillo, R.; Ozkurede, U.; Kim, Y.G.; Arindam, C.; Gale, M., Jr.; Silverman, R.H.; Colonna, M.; Akira, S.; et al. Cytosolic double-stranded RNA activates the NLRP3 inflammasome via MAVS-induced membrane permeabilization and K+ efflux. J. Immunol. 2014, 193, 4214–4222.
- Sheedy, F.J.; Grebe, A.; Rayner, K.J.; Kalantari, P.; Ramkhelawon, B.; Carpenter, S.B.; Becker, C.E.; Ediriweera, H.N.; Mullick, A.E.; Golenbock, D.T.; et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 2013, 14, 812–820.
- Chen, M.; Lu, X.; Lu, C.; Shen, N.; Jiang, Y.; Chen, M.; Wu, H. Soluble uric acid increases PDZK1 and ABCG2 expression in human intestinal cell lines via the TLR4-NLRP3 inflammasome and PI3K/Akt signaling pathway. Arthritis Res. Ther. 2018, 20, 20.
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411.
- Zhao, L.R.; Xing, R.L.; Wang, P.M.; Zhang, N.S.; Yin, S.J.; Li, X.C.; Zhang, L. NLRP1 and NLRP3 inflammasomes mediate LPS/ATPinduced pyroptosis in knee osteoarthritis. Mol. Med. Rep. 2018, 17, 5463–5469.
- Franchi, L.; Kanneganti, T.D.; Dubyak, G.R.; Nunez, G. Differential requirement of P2X7 receptor and intracellular K+ for caspase-1 activation induced by intracellular and extracellular bacteria. J. Biol. Chem. 2007, 282, 18810–18818.
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128.
- Kesavardhana, S.; Kanneganti, T.D. Mechanisms governing inflammasome activation, assembly and pyroptosis induction. Int. Immunol. 2017, 29, 201–210.
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254.
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665.
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116.
- Petrilli, V.; Papin, S.; Dostert, C.; Mayor, A.; Martinon, F.; Tschopp, J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007, 14, 1583–1589.
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414.
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225.
- Hornung, V.; Latz, E. Critical functions of priming and lysosomal damage for NLRP3 activation. Eur. J. Immunol. 2010, 40, 620–623.
- Rossol, M.; Pierer, M.; Raulien, N.; Quandt, D.; Meusch, U.; Rothe, K.; Schubert, K.; Schoneberg, T.; Schaefer, M.; Krugel, U.; et al. Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nat. Commun. 2012, 3, 1329.
- Locovei, S.; Wang, J.; Dahl, G. Activation of pannexin 1 channels by ATP through P2Y receptors and by cytoplasmic calcium. Febs Lett. 2006, 580, 239–244.
- He, Y.; Zeng, M.Y.; Yang, D.; Motro, B.; Nunez, G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016, 530, 354–357.
- Bauernfeind, F.; Bartok, E.; Rieger, A.; Franchi, L.; Nunez, G.; Hornung, V. Cutting edge: Reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J. Immunol. 2011, 187, 613–617.
- Juliana, C.; Fernandes-Alnemri, T.; Kang, S.; Farias, A.; Qin, F.; Alnemri, E.S. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J. Biol. Chem. 2012, 287, 36617–36622.
- Patel, M.N.; Carroll, R.G.; Galvan-Pena, S.; Mills, E.L.; Olden, R.; Triantafilou, M.; Wolf, A.I.; Bryant, C.E.; Triantafilou, K.; Masters, S.L. Inflammasome Priming in Sterile Inflammatory Disease. Trends Mol. Med. 2017, 23, 165–180.
- Stutz, A.; Kolbe, C.C.; Stahl, R.; Horvath, G.L.; Franklin, B.S.; van Ray, O.; Brinkschulte, R.; Geyer, M.; Meissner, F.; Latz, E. NLRP3 inflammasome assembly is regulated by phosphorylation of the pyrin domain. J. Exp. Med. 2017, 214, 1725–1736.
- Spalinger, M.R.; Kasper, S.; Gottier, C.; Lang, S.; Atrott, K.; Vavricka, S.R.; Scharl, S.; Raselli, T.; Frey-Wagner, I.; Gutte, P.M.; et al. NLRP3 tyrosine phosphorylation is controlled by protein tyrosine phosphatase PTPN22. J. Clin. Investig. 2016, 126, 1783–1800.
- Py, B.F.; Kim, M.S.; Vakifahmetoglu-Norberg, H.; Yuan, J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol. Cell 2013, 49, 331–338.
- Guo, C.; Xie, S.; Chi, Z.; Zhang, J.; Liu, Y.; Zhang, L.; Zheng, M.; Zhang, X.; Xia, D.; Ke, Y.; et al. Bile Acids Control Inflammation and Metabolic Disorder through Inhibition of NLRP3 Inflammasome. Immunity 2016, 45, 802–816.
- Neudecker, V.; Haneklaus, M.; Jensen, O.; Khailova, L.; Masterson, J.C.; Tye, H.; Biette, K.; Jedlicka, P.; Brodsky, K.S.; Gerich, M.E.; et al. Myeloid-derived miR-223 regulates intestinal inflammation via repression of the NLRP3 inflammasome. J. Exp. Med. 2017, 214, 1737–1752.
- Xie, Q.; Wei, M.; Zhang, B.; Kang, X.; Liu, D.; Zheng, W.; Pan, X.; Quan, Y.; Liao, D.; Shen, J. MicroRNA33 regulates the NLRP3 inflammasome signaling pathway in macrophages. Mol. Med. Rep. 2018, 17, 3318–3327.
- Rowczenio, D.M.; Gomes, S.M.; Arostegui, J.I.; Mensa-Vilaro, A.; Omoyinmi, E.; Trojer, H.; Baginska, A.; Baroja-Mazo, A.; Pelegrin, P.; Savic, S.; et al. Late-Onset Cryopyrin-Associated Periodic Syndromes Caused by Somatic NLRP3 Mosaicism-UK Single Center Experience. Front. Immunol. 2017, 8, 1410.
- Jiang, D.; Chen, S.; Sun, R.; Zhang, X.; Wang, D. The NLRP3 inflammasome: Role in metabolic disorders and regulation by metabolic pathways. Cancer Lett. 2018, 419, 8–19.
- He, Q.; Fu, Y.; Tian, D.; Yan, W. The contrasting roles of inflammasomes in cancer. Am. J. Cancer Res. 2018, 8, 566–583.
- Lang, Y.; Chu, F.; Shen, D.; Zhang, W.; Zheng, C.; Zhu, J.; Cui, L. Role of Inflammasomes in Neuroimmune and Neurodegenerative Diseases: A Systematic Review. Mediat. Inflamm. 2018, 2018, 1549549.
- Ott, A.; Stolk, R.; Van Harskamp, F.; Pols, H.; Hofman, A.; Breteler, M. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 1999, 53, 1937.
- Akter, K.; Lanza, E.A.; Martin, S.A.; Myronyuk, N.; Rua, M.; Raffa, R.B. Diabetes mellitus and Alzheimer’s disease: Shared pathology and treatment? Br. J. Clin. Pharmacol. 2011, 71, 365–376.
- Kroner, Z. The Relationship between Alzheimer’s Disease and Diabetes: Type 3 Diabetes. Altern. Med. Rev. 2009, 14.
- Ho, L.; Qin, W.; Pompl, P.N.; Xiang, Z.; Wang, J.; Zhao, Z.; Peng, Y.; Cambareri, G.; Rocher, A.; Mobbs, C.V. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. Faseb J. 2004, 18, 902–904.
- Jolivalt, C.G.; Hurford, R.; Lee, C.A.; Dumaop, W.; Rockenstein, E.; Masliah, E. Type 1 diabetes exaggerates features of Alzheimer’s disease in APP transgenic mice. Exp. Neurol. 2010, 223, 422–431.
- Takeda, S.; Sato, N.; Uchio-Yamada, K.; Sawada, K.; Kunieda, T.; Takeuchi, D.; Kurinami, H.; Shinohara, M.; Rakugi, H.; Morishita, R. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Aβ deposition in an Alzheimer mouse model with diabetes. Proc. Natl. Acad. Sci. USA 2010, 107, 7036–7041.
- Kanneganti, T.D.; Dixit, V.D. Immunological complications of obesity. Nat. Immunol. 2012, 13, 707–712.
- Donath, M.Y.; Shoelson, S.E. Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 2011, 11, 98–107.
- Scheen, A.J. Pathophysiology of type 2 diabetes. Acta Clin. Belg. 2003, 58, 335–341.
- Magliano, D.J.; Islam, R.M.; Barr, E.L.M.; Gregg, E.W.; Pavkov, M.E.; Harding, J.L.; Tabesh, M.; Koye, D.N.; Shaw, J.E. Trends in incidence of total or type 2 diabetes: Systematic review. Bmj 2019, 366, l5003.
- Henao-Mejia, J.; Elinav, E.; Thaiss, C.A.; Flavell, R.A. Inflammasomes and metabolic disease. Annu. Rev. Physiol. 2014, 76, 57–78.
- Eckel, R.H.; Kahn, S.E.; Ferrannini, E.; Goldfine, A.B.; Nathan, D.M.; Schwartz, M.W.; Smith, R.J.; Smith, S.R. Obesity and type 2 diabetes: What can be unified and what needs to be individualized? J. Clin. Endocrinol. Metab. 2011, 96, 1654–1663.
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808.
- Thomas, D.; Apovian, C. Macrophage functions in lean and obese adipose tissue. Metab. Clin. Exp. 2017, 72, 120–143.
- Francisco, V.; Pino, J.; Gonzalez-Gay, M.A.; Mera, A.; Lago, F.; Gomez, R.; Mobasheri, A.; Gualillo, O. Adipokines and inflammation: Is it a question of weight? Br. J. Pharmacol. 2018, 175, 1569–1579.
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188.
- Esser, N.; L’Homme, L.; De Roover, A.; Kohnen, L.; Scheen, A.J.; Moutschen, M.; Piette, J.; Legrand-Poels, S.; Paquot, N. Obesity phenotype is related to NLRP3 inflammasome activity and immunological profile of visceral adipose tissue. Diabetologia 2013, 56, 2487–2497.
- Yin, Z.; Deng, T.; Peterson, L.E.; Yu, R.; Lin, J.; Hamilton, D.J.; Reardon, P.R.; Sherman, V.; Winnier, G.E.; Zhan, M.; et al. Transcriptome analysis of human adipocytes implicates the NOD-like receptor pathway in obesity-induced adipose inflammation. Mol. Cell. Endocrinol. 2014, 394, 80–87.
- Bando, S.; Fukuda, D.; Soeki, T.; Nishimoto, S.; Uematsu, E.; Matsuura, T.; Ise, T.; Tobiume, T.; Yamaguchi, K.; Yagi, S.; et al. Expression of NLRP3 in subcutaneous adipose tissue is associated with coronary atherosclerosis. Atherosclerosis 2015, 242, 407–414.
- Kursawe, R.; Dixit, V.D.; Scherer, P.E.; Santoro, N.; Narayan, D.; Gordillo, R.; Giannini, C.; Lopez, X.; Pierpont, B.; Nouws, J.; et al. A Role of the Inflammasome in the Low Storage Capacity of the Abdominal Subcutaneous Adipose Tissue in Obese Adolescents. Diabetes 2016, 65, 610–618.
- Sokolova, M.; Sjaastad, I.; Louwe, M.C.; Alfsnes, K.; Aronsen, J.M.; Zhang, L.; Haugstad, S.B.; Bendiksen, B.A.; Ogaard, J.; Bliksoen, M.; et al. NLRP3 Inflammasome Promotes Myocardial Remodeling During Diet-Induced Obesity. Front. Immunol. 2019, 10, 1621.
- Nagareddy, P.R.; Kraakman, M.; Masters, S.L.; Stirzaker, R.A.; Gorman, D.J.; Grant, R.W.; Dragoljevic, D.; Hong, E.S.; Abdel-Latif, A.; Smyth, S.S.; et al. Adipose tissue macrophages promote myelopoiesis and monocytosis in obesity. Cell Metab. 2014, 19, 821–835.
- Mocanu, A.O.; Mulya, A.; Huang, H.; Dan, O.; Shimizu, H.; Batayyah, E.; Brethauer, S.A.; Dinischiotu, A.; Kirwan, J.P. Effect of Roux-en-Y Gastric Bypass on the NLRP3 Inflammasome in Adipose Tissue from Obese Rats. PLoS ONE 2015, 10, e0139764.
- Stienstra, R.; Tack, C.J.; Kanneganti, T.D.; Joosten, L.A.; Netea, M.G. The inflammasome puts obesity in the danger zone. Cell Metab. 2012, 15, 10–18.
- Rheinheimer, J.; de Souza, B.M.; Cardoso, N.S.; Bauer, A.C.; Crispim, D. Current role of the NLRP3 inflammasome on obesity and insulin resistance: A systematic review. Metab. Clin. Exp. 2017, 74, 1–9.
- Talukdar, S.; Oh, D.Y.; Bandyopadhyay, G.; Li, D.; Xu, J.; McNelis, J.; Lu, M.; Li, P.; Yan, Q.; Zhu, Y.; et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat. Med. 2012, 18, 1407–1412.
- Chawla, A.; Nguyen, K.D.; Goh, Y.P. Macrophage-mediated inflammation in metabolic disease. Nat. Rev. Immunol. 2011, 11, 738–749.
- Konner, A.C.; Bruning, J.C. Toll-like receptors: Linking inflammation to metabolism. Trends Endocrinol. Metab. 2011, 22, 16–23.
- Rogero, M.M.; Calder, P.C. Obesity, Inflammation, Toll-Like Receptor 4 and Fatty Acids. Nutrients 2018, 10, 432.
- Poggi, M.; Bastelica, D.; Gual, P.; Iglesias, M.A.; Gremeaux, T.; Knauf, C.; Peiretti, F.; Verdier, M.; Juhan-Vague, I.; Tanti, J.F.; et al. C3H/HeJ mice carrying a toll-like receptor 4 mutation are protected against the development of insulin resistance in white adipose tissue in response to a high-fat diet. Diabetologia 2007, 50, 1267–1276.
- Caricilli, A.M.; Nascimento, P.H.; Pauli, J.R.; Tsukumo, D.M.; Velloso, L.A.; Carvalheira, J.B.; Saad, M.J. Inhibition of toll-like receptor 2 expression improves insulin sensitivity and signaling in muscle and white adipose tissue of mice fed a high-fat diet. J. Endocrinol. 2008, 199, 399–406.
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184.
- Sokolova, M.; Ranheim, T.; Louwe, M.C.; Halvorsen, B.; Yndestad, A.; Aukrust, P. NLRP3 Inflammasome: A Novel Player in Metabolically Induced Inflammation-Potential Influence on the Myocardium. J. Cardiovasc. Pharmacol. 2019, 74, 276–284.
- Boni-Schnetzler, M.; Meier, D.T. Islet inflammation in type 2 diabetes. Semin. Immunopathol. 2019, 41, 501–513.
- Morikawa, S.; Kaneko, N.; Okumura, C.; Taguchi, H.; Kurata, M.; Yamamoto, T.; Osawa, H.; Nakanishi, A.; Zako, T.; Masumoto, J. IAPP/amylin deposition, which is correlated with expressions of ASC and IL-1beta in beta-cells of Langerhans’ islets, directly initiates NLRP3 inflammasome activation. Int. J. Immunopathol. Pharmacol. 2018, 32, 2058738418788749.
- Masters, S.L.; Dunne, A.; Subramanian, S.L.; Hull, R.L.; Tannahill, G.M.; Sharp, F.A.; Becker, C.; Franchi, L.; Yoshihara, E.; Chen, Z.; et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat. Immunol. 2010, 11, 897–904.
- Stienstra, R.; van Diepen, J.A.; Tack, C.J.; Zaki, M.H.; van de Veerdonk, F.L.; Perera, D.; Neale, G.A.; Hooiveld, G.J.; Hijmans, A.; Vroegrijk, I.; et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2011, 108, 15324–15329.
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.; Brickey, W.J.; Ting, J.P. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415.
- Tsalamandris, S.; Antonopoulos, A.S.; Oikonomou, E.; Papamikroulis, G.A.; Vogiatzi, G.; Papaioannou, S.; Deftereos, S.; Tousoulis, D. The Role of Inflammation in Diabetes: Current Concepts and Future Perspectives. Eur. Cardiol. 2019, 14, 50–59.
- Dinarello, C.A.; Donath, M.Y.; Mandrup-Poulsen, T. Role of IL-1beta in type 2 diabetes. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 314–321.
- Alexandraki, K.; Piperi, C.; Kalofoutis, C.; Singh, J.; Alaveras, A.; Kalofoutis, A. Inflammatory process in type 2 diabetes: The role of cytokines. Ann. N. Y. Acad. Sci. 2006, 1084, 89–117.
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dement. 2018, 4, 575–590.
- Qiu, C.; Kivipelto, M.; von Strauss, E. Epidemiology of Alzheimer’s disease: Occurrence, determinants, and strategies toward intervention. Dialogues Clin. Neurosci. 2009, 11, 111–128.
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356.
- Prinz, M.; Priller, J.; Sisodia, S.S.; Ransohoff, R.M. Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat. Neurosci. 2011, 14, 1227–1235.
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 2008, 9, 857–865.
- Nakanishi, A.; Kaneko, N.; Takeda, H.; Sawasaki, T.; Morikawa, S.; Zhou, W.; Kurata, M.; Yamamoto, T.; Akbar, S.M.F.; Zako, T.; et al. Amyloid beta directly interacts with NLRP3 to initiate inflammasome activation: Identification of an intrinsic NLRP3 ligand in a cell-free system. Inflamm. Regen. 2018, 38, 27.
- Ojala, J.; Alafuzoff, I.; Herukka, S.K.; van Groen, T.; Tanila, H.; Pirttila, T. Expression of interleukin-18 is increased in the brains of Alzheimer’s disease patients. Neurobiol. Aging 2009, 30, 198–209.
- Cartier, L.; Hartley, O.; Dubois-Dauphin, M.; Krause, K.H. Chemokine receptors in the central nervous system: Role in brain inflammation and neurodegenerative diseases. Brain Res. Brain Res. Rev. 2005, 48, 16–42.
- Lamkanfi, M.; Dixit, V.M. Inflammasomes and their roles in health and disease. Annu. Rev. Cell Dev. Biol. 2012, 28, 137–161.
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 2018, 17, 588–606.
- Lue, L.F.; Rydel, R.; Brigham, E.F.; Yang, L.B.; Hampel, H.; Murphy, G.M., Jr.; Brachova, L.; Yan, S.D.; Walker, D.G.; Shen, Y.; et al. Inflammatory repertoire of Alzheimer’s disease and nondemented elderly microglia in vitro. Glia 2001, 35, 72–79.
- Apelt, J.; Schliebs, R. Beta-amyloid-induced glial expression of both pro- and anti-inflammatory cytokines in cerebral cortex of aged transgenic Tg2576 mice with Alzheimer plaque pathology. Brain Res. 2001, 894, 21–30.
- Martinon, F.; Petrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241.
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678.
- Meyer-Luehmann, M.; Spires-Jones, T.L.; Prada, C.; Garcia-Alloza, M.; de Calignon, A.; Rozkalne, A.; Koenigsknecht-Talboo, J.; Holtzman, D.M.; Bacskai, B.J.; Hyman, B.T. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature 2008, 451, 720–724.
- Martini, A.C.; Gomez-Arboledas, A.; Forner, S.; Rodriguez-Ortiz, C.J.; McQuade, A.; Danhash, E.; Phan, J.; Javonillo, D.; Ha, J.V.; Tram, M.; et al. Amyloid-beta impairs TOM1-mediated IL-1R1 signaling. Proc. Natl. Acad. Sci. USA 2019, 116, 21198–21206.
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 2008, 28, 8354–8360.
- Ries, M.; Sastre, M. Mechanisms of Abeta Clearance and Degradation by Glial Cells. Front. Aging Neurosci. 2016, 8, 160.
- Varnum, M.M.; Ikezu, T. The classification of microglial activation phenotypes on neurodegeneration and regeneration in Alzheimer’s disease brain. Arch. Immunol. Ther. Exp. 2012, 60, 251–266.
- Baroja-Mazo, A.; Martin-Sanchez, F.; Gomez, A.I.; Martinez, C.M.; Amores-Iniesta, J.; Compan, V.; Barbera-Cremades, M.; Yague, J.; Ruiz-Ortiz, E.; Anton, J.; et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat. Immunol. 2014, 15, 738–748.
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived ASC specks cross-seed amyloid-beta in Alzheimer’s disease. Nature 2017, 552, 355–361.
- Morales, I.; Farias, G.; Maccioni, R.B. Neuroimmunomodulation in the pathogenesis of Alzheimer’s disease. Neuroimmunomodulation 2010, 17, 202–204.
- Wang, H.; Shen, Y.; Chuang, H.; Chiu, C.; Ye, Y.; Zhao, L. Neuroinflammation in Alzheimer’s Disease: Microglia, Molecular Participants and Therapeutic Choices. Curr. Alzheimer Res. 2019, 16, 659–674.
- Maccioni, R.B.; Rojo, L.E.; Fernandez, J.A.; Kuljis, R.O. The role of neuroimmunomodulation in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2009, 1153, 240–246.
- Griffin, W.S.; Liu, L.; Li, Y.; Mrak, R.E.; Barger, S.W. Interleukin-1 mediates Alzheimer and Lewy body pathologies. J. Neuroinflammation 2006, 3, 5.
- Biundo, F.; Del Prete, D.; Zhang, H.; Arancio, O.; D’Adamio, L. A role for tau in learning, memory and synaptic plasticity. Sci. Rep. 2018, 8, 3184.
- Murray, C.A.; Lynch, M.A. Evidence that increased hippocampal expression of the cytokine interleukin-1 beta is a common trigger for age- and stress-induced impairments in long-term potentiation. J. Neurosci. 1998, 18, 2974–2981.
- Kitazawa, M.; Cheng, D.; Tsukamoto, M.R.; Koike, M.A.; Wes, P.D.; Vasilevko, V.; Cribbs, D.H.; LaFerla, F.M. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal beta-catenin pathway function in an Alzheimer’s disease model. J. Immunol. 2011, 187, 6539–6549.
- Yin, J.; Zhao, F.; Chojnacki, J.E.; Fulp, J.; Klein, W.L.; Zhang, S.; Zhu, X. NLRP3 Inflammasome Inhibitor Ameliorates Amyloid Pathology in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 1977–1987.
- Chaparro-Huerta, V.; Rivera-Cervantes, M.C.; Flores-Soto, M.E.; Gomez-Pinedo, U.; Beas-Zarate, C. Proinflammatory cytokines and apoptosis following glutamate-induced excitotoxicity mediated by p38 MAPK in the hippocampus of neonatal rats. J. Neuroimmunol. 2005, 165, 53–62.
- Poddar, R.; Paul, S. Novel crosstalk between ERK MAPK and p38 MAPK leads to homocysteine-NMDA receptor-mediated neuronal cell death. J. Neurochem. 2013, 124, 558–570.
- Barone, F.C.; Irving, E.A.; Ray, A.M.; Lee, J.C.; Kassis, S.; Kumar, S.; Badger, A.M.; Legos, J.J.; Erhardt, J.A.; Ohlstein, E.H.; et al. Inhibition of p38 mitogen-activated protein kinase provides neuroprotection in cerebral focal ischemia. Med. Res. Rev. 2001, 21, 129–145.
- Serou, M.J.; DeCoster, M.A.; Bazan, N.G. Interleukin-1 beta activates expression of cyclooxygenase-2 and inducible nitric oxide synthase in primary hippocampal neuronal culture: Platelet-activating factor as a preferential mediator of cyclooxygenase-2 expression. J. Neurosci. Res. 1999, 58, 593–598.
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934.
- Batkulwar, K.; Godbole, R.; Banarjee, R.; Kassaar, O.; Williams, R.J.; Kulkarni, M.J. Advanced Glycation End Products Modulate Amyloidogenic APP Processing and Tau Phosphorylation: A Mechanistic Link between Glycation and the Development of Alzheimer’s Disease. ACS Chem. Neurosci. 2018, 9, 988–1000.
More
Information
Subjects:
Immunology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.4K
Revisions:
4 times
(View History)
Update Date:
25 May 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No