Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Pablo Hernansanz-Agustín | -- | 2462 | 2022-05-24 05:54:40 | | | |
| 2 | Vivi Li | Meta information modification | 2462 | 2022-05-24 06:10:28 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Hernansanz-Agustín, P.; Enríquez, J.A. Generation of Reactive Oxygen Species by Mitochondria. Encyclopedia. Available online: https://encyclopedia.pub/entry/23268 (accessed on 25 July 2026).
Hernansanz-Agustín P, Enríquez JA. Generation of Reactive Oxygen Species by Mitochondria. Encyclopedia. Available at: https://encyclopedia.pub/entry/23268. Accessed July 25, 2026.
Hernansanz-Agustín, Pablo, José Antonio Enríquez. "Generation of Reactive Oxygen Species by Mitochondria" Encyclopedia, https://encyclopedia.pub/entry/23268 (accessed July 25, 2026).
Hernansanz-Agustín, P., & Enríquez, J.A. (2022, May 24). Generation of Reactive Oxygen Species by Mitochondria. In Encyclopedia. https://encyclopedia.pub/entry/23268
Hernansanz-Agustín, Pablo and José Antonio Enríquez. "Generation of Reactive Oxygen Species by Mitochondria." Encyclopedia. Web. 24 May, 2022.
Copy Citation
Reactive oxygen species (ROS) are series of chemical products originated from one or several electron reductions of oxygen. ROS are involved in physiology and disease and can also be both cause and consequence of many biological scenarios. Mitochondria are the main source of ROS in the cell and, particularly, the enzymes in the electron transport chain are the major contributors to this phenomenon.
mitochondria
ROS
mechanism
signalling
disease
supercomplexes
1. Introduction
All metazoans require oxygen to survive as it is used by the mitochondria to obtain operational energy. Mitochondria are composed by a double membrane, the inner mitochondrial membrane (IMM) and the outer mitochondrial membrane (OMM), which are separated by the intermembrane space (IMS) and hold the interior of the organelle, the mitochondrial matrix, isolated form the cytoplasm. These organelles use a panoply of carbon sources, from catabolic to anabolic pathways, which generate reducing equivalents useful for the formation of adenosine 5′-triphosphate (ATP). Reducing equivalents, such as nicotinamide adenine dinucleotide hydrogen (NADH) or flavin adenine dinucleotide dihydrogen (FADH2), are co-factors in multiple reactions that store electrons derived from metabolic oxidations [1]. NADH flux independently and act as substrate of mitochondrial complex I (CI), which, in turn, reduces ubiquinone to ubiquinol. On the other hand, FADH2 remains linked to the enzymes that are in contact with the IMM, where they interact with CoQ to recycle their FAD cofactors by reducing ubiquinone to ubiquinol. The most prominent example of FAD-dependent enzymes in mitochondria is complex II (CII). Complex III (CIII) oxidizes ubiquinol to reduce cytochrome c, which, subsequently, donates its electron to complex IV (CIV). Finally, CIV reduces oxygen to water. CI, CIII, and CIV couple the electron flux to the ejection of H+ across the IMM, creating a H+ electrochemical gradient (negative and alkaline inside), the proton motive force (Δp). Δp is used by a fifth complex, the ATP synthase (CV), to transfer H+ back to the mitochondrial matrix in an energy-releasing process, which is used to phosphorylate adenosine 5′-diphosphate (ADP) into ATP. As long as the oxygen consumption matches the phosphorylation of ADP, the respiration is coupled to ATP synthesis. Complexes from CI to CIV comprise the mitochondrial electron transport chain (mETC), which, together with CV, form the oxidative phosphorylation system (OXPHOS; Figure 1) [1].
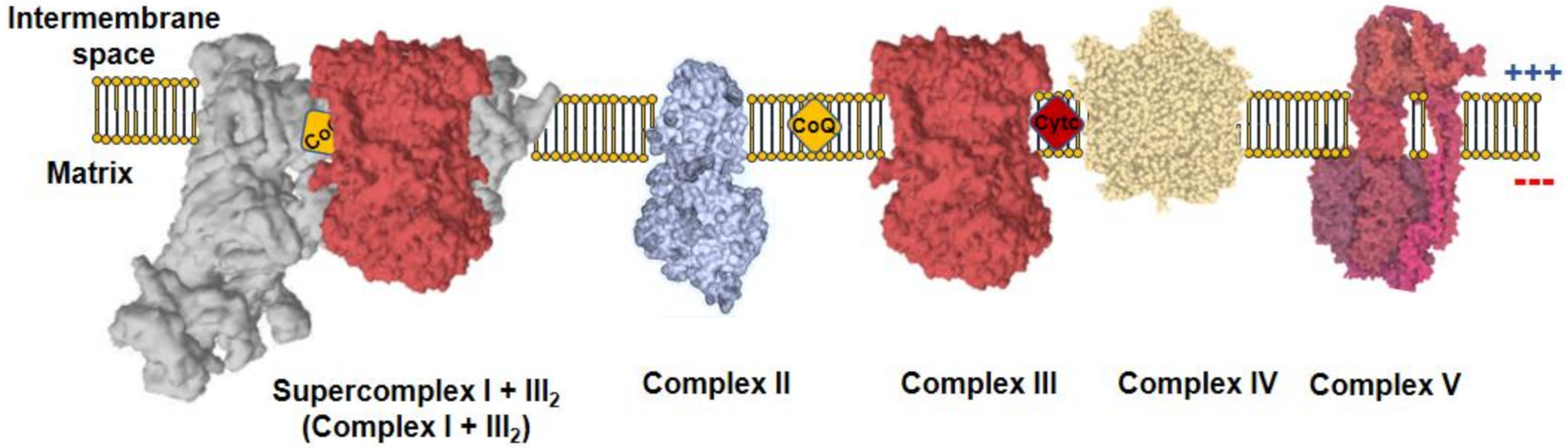
Figure 1. Mitochondrial oxidative phosphorylation system (OXPHOS). The inner mitochondrial membrane (IMM) comprises five protein complexes, which couple the transfer of electrons to H+ pumping. Charge distribution across the IMM produces a ΔΨm, negative inside. Complex I (CI) is normally found inside supercomplexes with complex III (CIII) (supercomplex I + III2) or CIII+complex IV (CIV) (N-respirasome), a particularly relevant feature for mitochondrial reactive oxygen species (ROS) production.
Δp is actively generated by the OXPHOS system; therefore, it reflects an active process of charge separation across the IMM, unlike the plasma membrane in which the charge separation is carried out by diffusion potential. Δp can be further decomposed in mitochondrial membrane potential (ΔΨm) and pH gradient (ΔpHm). ΔΨm is created by the difference in charge distribution across the IMM, being positive in the IMS and negative in the matrix. Similarly, the distribution of H+ across the IMM makes the IMS acidic and the matrix alkaline. An often-forgotten integral inner membrane protein that contribute to the H+ gradient is the NAD(P) transhydrogenase or NNT. NNT couples hydride transfer of reducing the equivalent between NADH and NADP to proton translocation across the inner mitochondrial membrane at the expenses of ∆p, generating NADPH of the proton gradient, but can work in reverse to generate ∆p and NADH from NADPH [2]. In addition to its role in ATP synthesis, the Δp is utilized by the mitochondria as the driving force to import proteins, metabolites and to balance the ion fluxes across the IMM. Interestingly, the movement of ions and charged molecules across the IMM impacts the Δp.
One of the more relevant ions that cross the IMM is Ca2+. Mitochondrial Ca2+ uptake is carried out by the inwardly rectifying mitochondrial Ca2+ uniporter (MCU), a highly specific channel for Ca2+ [3]. Thus, the MCU passes Ca2+ down the electrochemical gradient without coupling ATP hydrolysis or transport other ions. Mitochondrion Ca2+ extrusion is primarily carried out by the mitochondrial Na+/Ca2+ exchanger (NCLX) [4][5] in an electrophoretic process that mediates the extrusion of 3Na+ per 1Ca2+. The activity of NCLX depends on the Na+ gradient created by the fast acting mitochondrial Na+/H+ exchanger (mNHE), which parallels Na+ gradient to ΔpHm [6]. In this way, Ca2+ and Na+ homeostasis becomes engaged to the activity of OXPHOS. Notably, despite the importance of these exchangers for mitochondrial homeostasis, their molecular identities have remained unknown until recently [5][7][8], with the exception of the mNHE whose molecular identification remains to be confirmed.
All organisms are subjected to acute changes in oxygen availability (hyperoxia and hypoxia, respectively) and both result in the production of reactive oxygen species (ROS). ROS are most commonly the product of subsequent one-electron reduction of oxygen. Thus, one electron reduction of oxygen produces superoxide anion (O2•−), which is the most common first step in all ROS-producing enzymes and a very toxic species. One-electron reduction of O2•− produces hydrogen peroxide (H2O2), which is the best-known ROS acting as second messenger, normally due to its ability of reversibly oxidizing thiols groups on proteins. Subsequent one-electron reduction of H2O2 yields hydroxyl radical (•OH), which is an extremely harmful ROS that is notoriously involved in toxic reactions, such as the Fenton reaction. Mitochondrial ROS are involved in numerous physiological processes [9][10][11][12][13][14][15] and also link the progression from tissue homeostasis to disease [16][17][18][19]. Very surprisingly, the mechanisms of ROS production in live cells and tissues are poorly understood. Yet, thanks to the work on isolated mitochondria, researchers have a progressively deeper knowledge of how these organelles produce ROS [20][21][22][23][24].
2. Modes of ROS Production by Mitochondria
Classical experiments with mETC inhibitors pointed to CI and CIII as the major sources of ROS within the mitochondria and the cell. From a general point of view, potential sites of ROS production are triggered depending on the respiration substrate, membrane potential and, if present, the inhibitor used.
Under normal conditions, coupled respiration on glutamate/malate (GM) or pyruvate/malate (PM) activates the Krebs cycle enzymes 2-oxoglutarate dehydrogenase (OGDH), malate dehydrogenase (MDH), and pyruvate dehydrogenase (PDH), and maintains a low membrane potential as CV is producing ATP. OGDH, PDH, and MDH reduce NAD+ to NADH, which is, in turn, a substrate of CI. As electrons flow down the mETC they eventually reach CIII and CIV. In this situation, the production of ROS is low, but measurable and is commonly assigned to CI, the outer ubiquinone-binding site of CIII (CIIIo) and OGDH [22][23][24] in the so-called forward electron transfer (FET) (Figure 2A). This mode of ROS production can be exacerbated by ubiquinone-binding site inhibitors of CI, such as rotenone or piericidin A [25][26][27][28], or by inhibitors of the inner ubiquinone-binding site of CIII (CIIIi), such as antimicyn A [29][30]. On the other hand, to decrease this type of ROS production, especially after the application of the mentioned compounds, inhibitors of the flavin site of CI, such as diphenyleneiodonium (DPI; [31][32]), CIIIo blockers, such as myxothiazol, stigmatellin, or mucidin [33], and OGDH inhibitors, such as succinyl phosphonate or 3-methyl-2-oxopentanoate [24][34], can be applied.
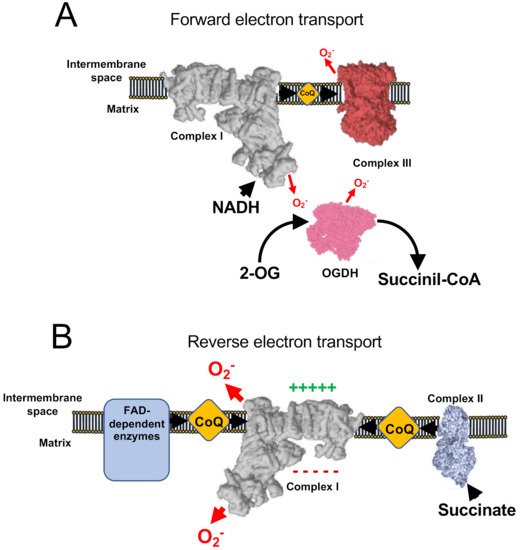
Figure 2. ROS production involving respiratory Complex I. (A) Under normal conditions, minimal amount of mitochondrial ROS are produced, nicotinamide adenine dinucleotide hydrogen (NADH) is oxidized at a high rate, as well as ubiquinol and 2-oxoglutarate. (B) Under conditions of normal-to-high ΔΨm and accumulation of reduced CoQ (or succinate) complex I works in the reverse mode, producing high rates of ROS. In addition, a few flavin adenine dinucleotide (FAD)-dependent enzymes have also been shown to contribute to reverse electron transfer (RET).
Succinate is the substrate of the Krebs cycle enzyme succinate dehydrogenase (SDH), also known as CII. SDH is representative of a variety of FAD dependent enzymes in the IMM that reduce ubiquinone, such as glyceraldehyde-3-phosphate dehydrogenase (G3PDH), dihydroorotate dehydrogenase (DHODH), and electron-transferring flavoprotein (ETF) [35]. When reducing potential is provided by CII, or to a lesser extent by other FAD-dependent enzymes, mitochondria over reduce ubiquinone and, under conditions of mitochondrial hyperpolarization (i.e., inactive CV), electrons are able to flow back through CI, reducing NAD+ into NADH and producing superoxide. This process is known as reverse electron transfer (RET) (Figure 2B). RET is the mode that produces the largest levels of ROS [36] and has been observed in both physiological [12][26] and pathophysiological situations [16][37][38]. Notably, the exact site of ROS production by RET is not yet clear as some authors propose that it occurs at the flavin site of CI (CIN; [36][39][40]), whereas others suggest the ubiquinone-binding site of CI (CIQ) plus CIN [41][42] or the iron-sulphur cluster N2 [43] as the main actors. The investigations of ROS production by RET have used a variety of commonly used mitochondrial drugs to halt this mechanism: (i) CI inhibitors such as rotenone, DPI [20][39] or piericidin A [42]; (ii) OXPHOS uncouplers, which are ΔΨm-disrupting molecules, such as FCCP [12][20][22][44]. As CI inhibitors can impede other modes of ROS production (see below), it is recommended to not only defining RET by its sensitivity to CI blockers (e.g., rotenone sensitivity), but also by using OXPHOS uncouplers. RET can be exacerbated by the incorporation of molecules to increase Δp [20], such as the CV inhibitor oligomycin or ATP, and it has been proposed that RET can be modulated by the activity of NNT as regulator of the NADH concentration [45][46]. Recently, it has been shown that mice harbouring ND6-P25L mutation in their mitochondrial DNA [47] are unable to produce rotenone-sensitive RET, though they still display some residual FCCP sensitivity [48]. Such inability is due to the capacity of CI to enter into its deactive form in every catalytic cycle, possibly allowing the enzyme to undergo active/deactive (A/D) transition very rapidly under hypoxia [48]. In addition, residual ROS production under succinate oxidation may be caused by the CIIIo [23][49].
As commented above, independently of the electron source, CIII is able to produce ROS upon inhibition with specific molecules. CIIIi inhibitors, such as Antimycin A promote the accumulation of electrons inside CIII, which reach the CIIIo site, producing superoxide anion. It is important to note that CIIIo blockers, such as myxothiazol or stigmatellin do not induce the production of ROS as they render the complex completely oxidized. Indeed, CIIIo inhibitors block the production of ROS by Antimycin A.
Under specific experimental conditions, when CI and CIII are inhibited and the concentration of succinate is low, CII can produce ROS at a significant rate (Figure 3). CII can also operate in forward and reverse modes as it can accept electrons both from succinate (forward) and ubiquinol (reverse). When working in the reverse mode (Figure 3A), CII can produce ROS sensitive to the ubiquinone-binding site inhibitor atpenin A5, and to the flavin-binding inhibitor malonate. However, when working on the forward mode (Figure 3B), CII can produce ROS that are only sensitive to monate, indicating that the site of ROS production under both modes is the flavin [49].
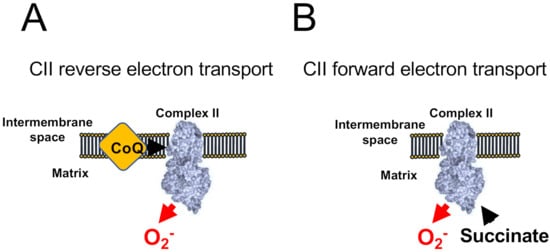
Figure 3. ROS production by Complex II. CII-dependent ROS production has been shown to occur only when CI and CIII are blocked. (A) When CoQ is highly reduced CII generates atpenin A5-sensitive ROS in its reverse reaction. (B) The accumulation of succinate promotes malonate-sensitive ROS in its forward reaction.
To note, ROS production triggered by oxidation of G3P can also be promoted by other sources, such as CI (RET) and, more scarcely, CIIIo [23][49]. In very specific experimental conditions, in which CI and CIII are inhibited, DHODH has been shown to produce ROS through CII and, to a lesser extent, by itself [50]. Moreover, OGDH, branched-chain 2-oxoacid dehydrogenase (BCODH) and pyruvate dehydrogenase (PDH) are potential sources of ROS under precise experimental conditions (Table 1) [34].
Table 1. Summary of the main ROS sources in mitochondria. Respiratory enzymes are able to produce ROS in forward and reverse reactions, as well as under specific physiological conditions, such during acute hypoxia. Biological material is underlined.
| Name and Source | System | Substrate(s)/Conditions | Potentiator(s) | Inhibitor(s) | References |
|---|---|---|---|---|---|
| FET (CI and CIIIo) | Tissues, cells, and isolated mitochondria | Cells and Tissues: standard culture media Isolated Mitochondria: pyruvate, malate and glutamate |
Rotenone, Piericidin A or Antimycin A | DPI, myxothiazol, stigmatellin or mucidin | [25][26][28][29][30][31][32][33][34] |
| RET (CIN) | Cells and isolated mitochondria | Succinate or G3P | CV inhibitors or ATP | CI and CII inhibitors and OXPHOS uncouplers (FCCP) | [13][16][20][22][36][37][38][39][40][41][42][44] |
| CIIIo | Cells and isolated mitochondria | NADH or succinate | Antimycin A | Myxothiazol or stigmatellin | [29][30][51] |
| CII-derived forward ROS production | Isolated mitochondria | Low succinate concentration, CI and CIII inhibited | - | Malonate | [49] |
| CII-derived reverse ROS production | Isolated mitochondria | Ubiquinol concentration, CI and CIII inhibited | - | Atpenin A5 and malonate | [49] |
| Hypoxic ROS | Tissues, cells and isolated mitochondria | Cells and Tissues: normal culture media Isolated Mitochondria: Malate, glutamate, CaCl2 and NaCl |
Monensin, Nigericin, FCCP (in normoxic cells) | Rotenone, piericidin A, myxothiazol, malonate, NCLX inhibitors (preincubated) | [52] |
3. ROS in Acute Hypoxia
Hypoxia is defined as the decreased availability of oxygen in cells and tissues. Low oxygen levels trigger a series of responses that are related to many physiological and pathophysiological scenarios [53]. The counterintuitive observation that hypoxia induced the production of ROS was called the ROS paradox in hypoxia [54][55]. Nowadays, though the relationship between chronic adaptation to hypoxia and the production of ROS is still unclear, acute responses to hypoxia is undoubtedly associated to hypoxic ROS generation [52][56][57][58][59][60].
During the first minutes of hypoxia, CI undergoes the active/deactive (A/D) transition, which consists of a conformational change involving ND3 and other CI subunits. ND3 rearranges to expose its Cys39 [61][62][63][64]. A/D transition is a characteristic dormant state of CI that is not able to perform its enzymatic activity and, therefore, is not able to pump H+ [27][65]. As CI becomes deactive in acute hypoxia, the mitochondrial matrix acidifies and the calcium phosphate precipitates in the matrix partially dissolve, liberating free Ca2+. The rise in matrix [Ca2+] activates the mitochondrial Ca2+/Na+ antiporter (NCLX), which promotes the entry of Na+ into the mitochondria. Na+ accumulates in the matrix and interacts with the carbonyl group in the phospholipids of the inner leaflet of the IMM. The interaction Na+:phospholipid promotes the formation of phospholipid aggregates, which, in turn, diminish the fluidity of IMM. The decrease in IMM fluidity lowers ubiquinol transfer between CII and CIII, while the transfer between CI and CIII is preserved as they are arranged into supercomplexes [66][67]. The decrease in ubiquinol transfer between CII and CIII promotes the generation of superoxide at the level of CIIIo due to the uncoupling of the Q cycle. This mechanism highlights the role of Na+ as second messenger, controlling OXPHOS and hypoxic redox signalling [52].
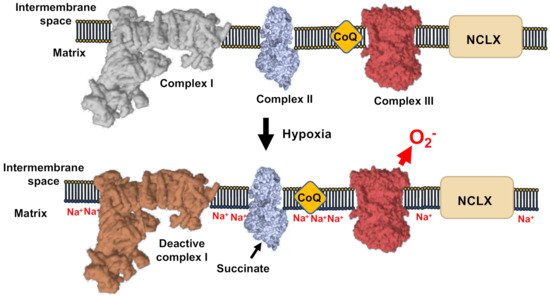
Figure 4. Mechanism of ROS production in hypoxia. CI-deactivation upon oxygen reduction induce de acidification of the mitochondrial matrix, the subsequent solubilization of matrix Ca2+ precipitates. The concomitant elevation of Ca2+ concentration activate the Ca2+/Na+ antiporter (NCLX). This causes the elevation of Na+ that decrease the inner membrane fluidity affecting the free CoQ diffusion and increase ROS production by CIII.
References
- Enriquez, J.A. Supramolecular Organization of Respiratory Complexes. Annu. Rev. Physiol. 2016, 78, 533–561.
- Kampjut, D.; Sazanov, L.A. Structure and mechanism of mitochondrial proton-translocating transhydrogenase. Nature 2019, 573, 291–295.
- Kirichok, Y.; Krapivinsky, G.; Clapham, D.E. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 2004, 427, 360–364.
- De Marchi, U.; Santo-Domingo, J.; Castelbou, C.; Sekler, I.; Wiederkehr, A.; Demaurex, N. NCLX protein, but not LETM1, mediates mitochondrial Ca2+ extrusion, thereby limiting Ca2+-induced NAD(P)H production and modulating matrix redox state. J. Biol. Chem. 2014, 289, 20377–20385.
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441.
- Jung, D.W.; Apel, L.M.; Brierley, G.P. Transmembrane gradients of free Na+ in isolated heart mitochondria estimated using a fluorescent probe. Am. J. Physiol. 1992, 262, C1047–C1055.
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345.
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabo, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340.
- Acin-Perez, R.; Iborra, S.; Marti-Mateos, Y.; Cook, E.C.L.; Conde-Garrosa, R.; Petcherski, A.; Munoz, M.D.M.; Martinez de Mena, R.; Krishnan, K.C.; Jimenez, C.; et al. Fgr kinase is required for proinflammatory macrophage activation during diet-induced obesity. Nat. Metab. 2020, 2, 974–988.
- Gorlach, A.; Dimova, E.Y.; Petry, A.; Martinez-Ruiz, A.; Hernansanz-Agustin, P.; Rolo, A.P.; Palmeira, C.M.; Kietzmann, T. Reactive oxygen species, nutrition, hypoxia and diseases: Problems solved? Redox Biol. 2015, 6, 372–385.
- Latorre-Pellicer, A.; Moreno-Loshuertos, R.; Lechuga-Vieco, A.V.; Sanchez-Cabo, F.; Torroja, C.; Acin-Perez, R.; Calvo, E.; Aix, E.; Gonzalez-Guerra, A.; Logan, A.; et al. Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature 2016, 535, 561–565.
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Dabritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457–470.e13.
- Mills, E.L.; Pierce, K.A.; Jedrychowski, M.P.; Garrity, R.; Winther, S.; Vidoni, S.; Yoneshiro, T.; Spinelli, J.B.; Lu, G.Z.; Kazak, L.; et al. Accumulation of succinate controls activation of adipose tissue thermogenesis. Nature 2018, 560, 102–106.
- Moreno-Loshuertos, R.; Acin-Perez, R.; Fernandez-Silva, P.; Movilla, N.; Perez-Martos, A.; Rodriguez de Cordoba, S.; Gallardo, M.E.; Enriquez, J.A. Differences in reactive oxygen species production explain the phenotypes associated with common mouse mitochondrial DNA variants. Nat. Genet. 2006, 38, 1261–1268.
- Vicente-Gutierrez, C.; Bonora, N.; Bobo-Jimenez, V.; Jimenez-Blasco, D.; Lopez-Fabuel, I.; Fernandez, E.; Josephine, C.; Bonvento, G.; Enriquez, J.A.; Almeida, A.; et al. Astrocytic mitochondrial ROS modulate brain metabolism and mouse behaviour. Nat. Metab. 2019, 1, 201–211.
- Chouchani, E.T.; Pell, V.R.; James, A.M.; Work, L.M.; Saeb-Parsy, K.; Frezza, C.; Krieg, T.; Murphy, M.P. A Unifying Mechanism for Mitochondrial Superoxide Production during Ischemia-Reperfusion Injury. Cell Metab. 2016, 23, 254–263.
- Guzman, J.N.; Sanchez-Padilla, J.; Wokosin, D.; Kondapalli, J.; Ilijic, E.; Schumacker, P.T.; Surmeier, D.J. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 2010, 468, 696–700.
- Harris, I.S.; DeNicola, G.M. The Complex Interplay between Antioxidants and ROS in Cancer. Trends Cell Biol. 2020, 30, 440–451.
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167.
- Drose, S.; Brandt, U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv. Exp. Med. Biol. 2012, 748, 145–169.
- Drose, S.; Galkin, A.; Brandt, U. Chapter 26 Measurement of superoxide formation by mitochondrial complex I of Yarrowia lipolytica. Methods Enzymol. 2009, 456, 475–490.
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13.
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013, 1, 304–312.
- Wong, H.S.; Dighe, P.A.; Mezera, V.; Monternier, P.A.; Brand, M.D. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 2017, 292, 16804–16809.
- Chernyak, B.V.; Izyumov, D.S.; Lyamzaev, K.G.; Pashkovskaya, A.A.; Pletjushkina, O.Y.; Antonenko, Y.N.; Sakharov, D.V.; Wirtz, K.W.; Skulachev, V.P. Production of reactive oxygen species in mitochondria of HeLa cells under oxidative stress. Biochim. Biophys. Acta 2006, 1757, 525–534.
- Guaras, A.; Perales-Clemente, E.; Calvo, E.; Acin-Perez, R.; Loureiro-Lopez, M.; Pujol, C.; Martinez-Carrascoso, I.; Nunez, E.; Garcia-Marques, F.; Rodriguez-Hernandez, M.A.; et al. The CoQH2/CoQ Ratio Serves as a Sensor of Respiratory Chain Efficiency. Cell Rep. 2016, 15, 197–209.
- Hernansanz-Agustin, P.; Ramos, E.; Navarro, E.; Parada, E.; Sanchez-Lopez, N.; Pelaez-Aguado, L.; Cabrera-Garcia, J.D.; Tello, D.; Buendia, I.; Marina, A.; et al. Mitochondrial complex I deactivation is related to superoxide production in acute hypoxia. Redox Biol. 2017, 12, 1040–1051.
- Korge, P.; Calmettes, G.; Weiss, J.N. Reactive oxygen species production in cardiac mitochondria after complex I inhibition: Modulation by substrate-dependent regulation of the NADH/NAD(+) ratio. Free Radic. Biol. Med. 2016, 96, 22–33.
- Hernansanz-Agustin, P.; Choya-Foces, C.; Martinez-Ruiz, A. Measurement of Superoxide Production in Acute Hypoxia by Fixed-Cell Microscopy. Methods Mol. Biol. 2021, 2202, 43–50.
- Hernansanz-Agustin, P.; Izquierdo-Alvarez, A.; Sanchez-Gomez, F.J.; Ramos, E.; Villa-Pina, T.; Lamas, S.; Bogdanova, A.; Martinez-Ruiz, A. Acute hypoxia produces a superoxide burst in cells. Free Radic. Biol. Med. 2014, 71, 146–156.
- Gusdon, A.M.; Fernandez-Bueno, G.A.; Wohlgemuth, S.; Fernandez, J.; Chen, J.; Mathews, C.E. Respiration and substrate transport rates as well as reactive oxygen species production distinguish mitochondria from brain and liver. BMC Biochem. 2015, 16, 22.
- Liu, Y.; Fiskum, G.; Schubert, D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J. Neurochem. 2002, 80, 780–787.
- Fato, R.; Bergamini, C.; Bortolus, M.; Maniero, A.L.; Leoni, S.; Ohnishi, T.; Lenaz, G. Differential effects of mitochondrial Complex I inhibitors on production of reactive oxygen species. Biochim. Biophys. Acta 2009, 1787, 384–392.
- Quinlan, C.L.; Goncalves, R.L.; Hey-Mogensen, M.; Yadava, N.; Bunik, V.I.; Brand, M.D. The 2-oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. J. Biol. Chem. 2014, 289, 8312–8325.
- Scialo, F.; Fernandez-Ayala, D.J.; Sanz, A. Role of Mitochondrial Reverse Electron Transport in ROS Signaling: Potential Roles in Health and Disease. Front. Physiol. 2017, 8, 428.
- Robb, E.L.; Hall, A.R.; Prime, T.A.; Eaton, S.; Szibor, M.; Viscomi, C.; James, A.M.; Murphy, M.P. Control of mitochondrial superoxide production by reverse electron transport at complex I. J. Biol. Chem. 2018, 293, 9869–9879.
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435.
- Scialo, F.; Sriram, A.; Fernandez-Ayala, D.; Gubina, N.; Lohmus, M.; Nelson, G.; Logan, A.; Cooper, H.M.; Navas, P.; Enriquez, J.A.; et al. Mitochondrial ROS Produced via Reverse Electron Transport Extend Animal Lifespan. Cell Metab. 2016, 23, 725–734.
- Pryde, K.R.; Hirst, J. Superoxide is produced by the reduced flavin in mitochondrial complex I: A single, unified mechanism that applies during both forward and reverse electron transfer. J. Biol. Chem. 2011, 286, 18056–18065.
- Stepanova, A.; Kahl, A.; Konrad, C.; Ten, V.; Starkov, A.S.; Galkin, A. Reverse electron transfer results in a loss of flavin from mitochondrial complex I: Potential mechanism for brain ischemia reperfusion injury. J. Cereb. Blood Flow Metab. 2017, 37, 3649–3658.
- Brand, M.D.; Goncalves, R.L.; Orr, A.L.; Vargas, L.; Gerencser, A.A.; Borch Jensen, M.; Wang, Y.T.; Melov, S.; Turk, C.N.; Matzen, J.T.; et al. Suppressors of Superoxide-H2O2 Production at Site IQ of Mitochondrial Complex I Protect against Stem Cell Hyperplasia and Ischemia-Reperfusion Injury. Cell Metab. 2016, 24, 582–592.
- Ohnishi, S.T.; Ohnishi, T.; Muranaka, S.; Fujita, H.; Kimura, H.; Uemura, K.; Yoshida, K.; Utsumi, K. A possible site of superoxide generation in the complex I segment of rat heart mitochondria. J. Bioenerg. Biomembr. 2005, 37, 1–15.
- Genova, M.L.; Ventura, B.; Giuliano, G.; Bovina, C.; Formiggini, G.; Parenti Castelli, G.; Lenaz, G. The site of production of superoxide radical in mitochondrial Complex I is not a bound ubisemiquinone but presumably iron-sulfur cluster N2. FEBS Lett. 2001, 505, 364–368.
- Komlodi, T.; Geibl, F.F.; Sassani, M.; Ambrus, A.; Tretter, L. Membrane potential and delta pH dependency of reverse electron transport-associated hydrogen peroxide production in brain and heart mitochondria. J. Bioenerg. Biomembr. 2018, 50, 355–365.
- Albracht, S.P.; Meijer, A.J.; Rydstrom, J. Mammalian NADH:ubiquinone oxidoreductase (Complex I) and nicotinamide nucleotide transhydrogenase (Nnt) together regulate the mitochondrial production of H(2)O(2)--implications for their role in disease, especially cancer. J. Bioenerg. Biomembr. 2011, 43, 541–564.
- Sharaf, M.S.; Stevens, D.; Kamunde, C. Mitochondrial transition ROS spike (mTRS) results from coordinated activities of complex I and nicotinamide nucleotide transhydrogenase. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 955–965.
- Lin, C.S.; Sharpley, M.S.; Fan, W.; Waymire, K.G.; Sadun, A.A.; Carelli, V.; Ross-Cisneros, F.N.; Baciu, P.; Sung, E.; McManus, M.J.; et al. Mouse mtDNA mutant model of Leber hereditary optic neuropathy. Proc. Natl. Acad. Sci. USA 2012, 109, 20065–20070.
- Yin, Z.; Burger, N.; Kula-Alwar, D.; Aksentijevic, D.; Bridges, H.R.; Prag, H.A.; Grba, D.N.; Viscomi, C.; James, A.M.; Mottahedin, A.; et al. Structural basis for a complex I mutation that blocks pathological ROS production. Nat. Commun. 2021, 12, 707.
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J. Biol. Chem. 2012, 287, 27255–27264.
- Hey-Mogensen, M.; Goncalves, R.L.; Orr, A.L.; Brand, M.D. Production of superoxide/H2O2 by dihydroorotate dehydrogenase in rat skeletal muscle mitochondria. Free Radic. Biol. Med. 2014, 72, 149–155.
- Drose, S.; Brandt, U. The mechanism of mitochondrial superoxide production by the cytochrome bc1 complex. J. Biol. Chem. 2008, 283, 21649–21654.
- Hernansanz-Agustin, P.; Choya-Foces, C.; Carregal-Romero, S.; Ramos, E.; Oliva, T.; Villa-Pina, T.; Moreno, L.; Izquierdo-Alvarez, A.; Cabrera-Garcia, J.D.; Cortes, A.; et al. Na(+) controls hypoxic signalling by the mitochondrial respiratory chain. Nature 2020, 586, 287–291.
- Lopez-Barneo, J.; Simon, M.C. Cellular adaptation to oxygen deficiency beyond the Nobel award. Nat. Commun. 2020, 11, 607.
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720.
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005, 1, 401–408.
- Arias-Mayenco, I.; Gonzalez-Rodriguez, P.; Torres-Torrelo, H.; Gao, L.; Fernandez-Aguera, M.C.; Bonilla-Henao, V.; Ortega-Saenz, P.; Lopez-Barneo, J. Acute O2 Sensing: Role of Coenzyme QH2/Q Ratio and Mitochondrial ROS Compartmentalization. Cell Metab. 2018, 28, 145–158.e144.
- Fernandez-Aguera, M.C.; Gao, L.; Gonzalez-Rodriguez, P.; Pintado, C.O.; Arias-Mayenco, I.; Garcia-Flores, P.; Garcia-Perganeda, A.; Pascual, A.; Ortega-Saenz, P.; Lopez-Barneo, J. Oxygen Sensing by Arterial Chemoreceptors Depends on Mitochondrial Complex I Signaling. Cell Metab. 2015, 22, 825–837.
- Paddenberg, R.; Ishaq, B.; Goldenberg, A.; Faulhammer, P.; Rose, F.; Weissmann, N.; Braun-Dullaeus, R.C.; Kummer, W. Essential role of complex II of the respiratory chain in hypoxia-induced ROS generation in the pulmonary vasculature. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L710–L719.
- Smith, K.A.; Schumacker, P.T. Sensors and signals: The role of reactive oxygen species in hypoxic pulmonary vasoconstriction. J. Physiol. 2019, 597, 1033–1043.
- Waypa, G.B.; Smith, K.A.; Schumacker, P.T. O2 sensing, mitochondria and ROS signaling: The fog is lifting. Mol. Aspects Med. 2016, 47–48, 76–89.
- Agip, A.A.; Blaza, J.N.; Bridges, H.R.; Viscomi, C.; Rawson, S.; Muench, S.P.; Hirst, J. Cryo-EM structures of complex I from mouse heart mitochondria in two biochemically defined states. Nat. Struct. Mol. Biol. 2018, 25, 548–556.
- Blaza, J.N.; Vinothkumar, K.R.; Hirst, J. Structure of the Deactive State of Mammalian Respiratory Complex I. Structure 2018, 26, 312–319.e313.
- Galkin, A.; Meyer, B.; Wittig, I.; Karas, M.; Schagger, H.; Vinogradov, A.; Brandt, U. Identification of the mitochondrial ND3 subunit as a structural component involved in the active/deactive enzyme transition of respiratory complex I. J. Biol. Chem. 2008, 283, 20907–20913.
- Kampjut, D.; Sazanov, L.A. The coupling mechanism of mammalian respiratory complex I. Science 2020, 370, eabc4209.
- Drose, S.; Stepanova, A.; Galkin, A. Ischemic A/D transition of mitochondrial complex I and its role in ROS generation. Biochim. Biophys. Acta 2016, 1857, 946–957.
- Calvo, E.; Cogliati, S.; Hernansanz-Agustin, P.; Loureiro-Lopez, M.; Guaras, A.; Casuso, R.A.; Garcia-Marques, F.; Acin-Perez, R.; Marti-Mateos, Y.; Silla-Castro, J.C.; et al. Functional role of respiratory supercomplexes in mice: SCAF1 relevance and segmentation of the Qpool. Sci. Adv. 2020, 6, eaba7509.
- Lapuente-Brun, E.; Moreno-Loshuertos, R.; Acin-Perez, R.; Latorre-Pellicer, A.; Colas, C.; Balsa, E.; Perales-Clemente, E.; Quiros, P.M.; Calvo, E.; Rodriguez-Hernandez, M.A.; et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 2013, 340, 1567–1570.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.7K
Revisions:
2 times
(View History)
Update Date:
24 May 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No