Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Marc Vervloet | -- | 1907 | 2022-05-16 10:37:26 | | | |
| 2 | Dean Liu | Meta information modification | 1907 | 2022-05-17 07:56:04 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Vervloet, M. Shedding Light on the Complex Regulation of FGF23. Encyclopedia. Available online: https://encyclopedia.pub/entry/22960 (accessed on 27 June 2026).
Vervloet M. Shedding Light on the Complex Regulation of FGF23. Encyclopedia. Available at: https://encyclopedia.pub/entry/22960. Accessed June 27, 2026.
Vervloet, Marc. "Shedding Light on the Complex Regulation of FGF23" Encyclopedia, https://encyclopedia.pub/entry/22960 (accessed June 27, 2026).
Vervloet, M. (2022, May 16). Shedding Light on the Complex Regulation of FGF23. In Encyclopedia. https://encyclopedia.pub/entry/22960
Vervloet, Marc. "Shedding Light on the Complex Regulation of FGF23." Encyclopedia. Web. 16 May, 2022.
Copy Citation
Early research has suggested a rather straightforward relation between phosphate exposure, increased serum FGF23 (Fibroblast Growth Factor 23) concentrations and clinical endpoints.
FGF23 (Fibroblast Growth Factor 23)
regulation
mineral metabolism
1. Introduction
FGF23 is a hormone, secreted by osteocytes, and has a central physiological role in phosphate homeostasis. It promotes phosphaturia and inhibits the activation of vitamin D, thereby limiting vitamin-D mediated phosphate absorption from the diet by the transcellular uptake route of enterocytes in the gastro-intestinal tract. There are several principal ways in which FGF23 concentrations can be regulated, and all of these appear to play a role. These modes of regulation are production and secretion by the cells of origin, ectopic production, and cleavage or breakdown at cells of origin or after release into the circulation. The currently available immunoassays measure either the full-length and biologically active hormone, termed intact FGF23 (iFGF23), or both iFGF23 and its c-terminal fragment, termed cFGF23. It should be noted that the term cFGF23 for this assay is a confusing term, because it does not only measure the c-terminal fragment, the latter originating after cleavage of the full-length polypeptide. This cleavage obviously also generates an N-terminal fragment, but no commercially available assay detects this fraction.
2. The Role of Minerals as Regulators of FGF23
2.1. Phosphate
Given the key role of FGF23 to protect the organism against hyperphosphatemia, it can be expected that phosphate increases FGF23 concentrations. Indeed, several studies have shown that an increase in dietary intake of phosphate by both healthy volunteers and people with CKD increased its concentrations, albeit with some delay of around 24 h [1][2][3] to restore phosphate balance. In turn, phosphate restriction has the ability to lower FGF23, but, different from PTH secretion from healthy parathyroid glands in a setting of hypercalcemia, FGF23 has never been described to be fully suppressed following hypophosphatemia, for instance when induced by mutations in the renal phosphate transporter NaPi2c which gives rise to hereditary hypophosphatemic rickets with hypercalciuria (HHRH) [4][5]. Until recently, the underlying molecular mechanism by which phosphate modulates FGF23 levels has been elusive. It has been shown that phosphate transport into bone cells across the inorganic phosphate transporter 1 (PiT-1) may be involved [6]. A recent study revealed an additional remarkable mechanism [7]. Bone cells that produce FGF23 express its receptor FGFR1 as well. It is now shown that phosphate itself can bind to this unliganded receptor, leading to the upregulation of the Galnt3 gene, the protein-product of which leads to O-glycosylation of full-length FGF23, as will be discussed below. The consequence of this post-translational modification of the FGF23 molecule is that it escapes intracellular cleavage, increasing the proportion of biologically active FGF23. This mechanism does not suggest that phosphate induces FGF23 expression, even though a previous study suggested it can in a cell line [8], but rather stabilises the hormone. This mode of action of phosphate on FGF23 concentrations is in line with clinical studies in patients with CKD that addressed the question of whether dietary phosphate restriction can lower FGF23. A recent meta-analysis of these studies found more pronounced reduction of iFGF23 than of cFGF23, the latter also measuring FGF23 fragments [9]. In normophosphatemic CKD patients, short-term treatment with non-calcium containing phosphate binders did not change FGF23 [10][11], while prolonged treatment induced a substantial decline [12]. The use of calcium-containing binders did increase FGF23 [13].
2.2. Calcium
Interestingly, there appears to be a minimal concentration of calcium required for phosphate to be able to increase FGF23 levels. In an animal model testing varying serum concentration of calcium, it was shown that an increment of FGF23 by PTH was completely abolished when ionized calcium concentrations were below 4 mg/dL [14]. The physiological functionality of this phenomenon might be that this prevents the catabolism of vitamin D by FGF23 in a setting of hypocalcemia. Moreover, in an animal model, calcium itself was shown to be able to directly increase FGF23 transcription by acting on the promotor of the Fgf23 gene [15][16]. These findings from experimental research are in line with most, but not all, clinical observations. In a clinical trial among 30 early CKD patients, studying the effects of adding calcium carbonate to calcitriol, it was shown that this induced an increase of FGF23, which was paralleled by an increase in serum calcium concentration [17]. In more advanced CKD, the non-calcium containing phosphate binder lanthanum carbonate was able to lower FGF23 levels, while a calcium-containing binder could not [13]. However, in a short-term study, acute increments or decrements of serum calcium concentrations had no effect on FGF23 [18].
2.3. Calciprotein Particles
Apart from the synergistic effects of combined higher levels of calcium and phosphate on increasing FGF23, it is possible that this is mediated by the formation of calciprotein particles (CPP) [19][20]. Even at physiological concentrations, human plasma is supersaturated for calcium and phosphate, which would induce spontaneous hydroxyapatite crystal formation [21]. These potentially damaging crystals, however, are prevented from being formed and freely floating in the circulation by being scavenged into soluble amorphous primary calciprotein particles CPP (CPP1), which are nanoparticles containing the serum protein Fetuin-A as the main protein constituent. In a setting of increased availability of these minerals, as is the case for phosphate in CKD, or suppressed hepatic production of Fetuin A in a setting of chronic inflammation, the stage is set to overwhelm the capacity of this defence system, leading to the formation of more toxic larger crystalline secondary CPPs (CPP2) [22][23][24]. Like high exposure to phosphate, exposure to high calcium levels also increases the amount of CPP, as was shown in a patient with renal sarcoidosis, and this was paralleled by an increase in FGF23 [25]. The role of calcium on the formation of CPP was also shown in a clinical study comparing calciumcarbonate with lanthanumcarbonate [26]. After switching to lanthanumcarbonate, the total amount of CPP declined substantially, without major differences in serum concentration of calcium and phosphate between the two phosphate binders.
A recent clinical observational study demonstrated an association between the amount of CPPs and FGF23, suggesting an induction in the phosphaturic hormone by CPP’s [27]. Indeed, a recent in vitro study found that CPPs are capable of increasing FGF23 expression in osteoblast-like cells [28]. Remarkably, this effect was restricted to the smaller sized CPP1. It is therefore conceivable that an increased amount of CPP’s formed triggers FGF23, which in turn induces phosphaturia and declines levels of active vitamin D. FGF23 thereby slows the formation of CPP’s by lowering the concentrations of the minerals that form its mineral content. This concept is supported by the ability of CPP to exit the circulation, enter the bone marrow and reach FGF23-producing bone cells [28].
2.4. Magnesium
Given its resemblance to calcium as a bivalent cation, and its beneficial effects on the formation of CPP [29][30][31], it is likely that magnesium is also involved in the regulation of FGF23. Data, however, are scarce. In an animal study of cats with chronic kidney disease, a negative association between serum magnesium concentration and FGF23 was found, which was independent of calcium, phosphate, and PTH [32]. In an observational study among young healthy men, it was shown that a lower dietary intake of magnesium was associated with higher FGF23 [33]. When rats were exposed to a short-term (7 day) magnesium deficient diet, FGF23 levels were higher compared to a normal diet at all time points following the interventions which reached statistical significance after one week [34]. However, clinical trials demonstrating a beneficial effect on clinically relevant endpoints of magnesium supplements are lacking [35].
The role of minerals and calciprotein particles are summarized in Figure 1.
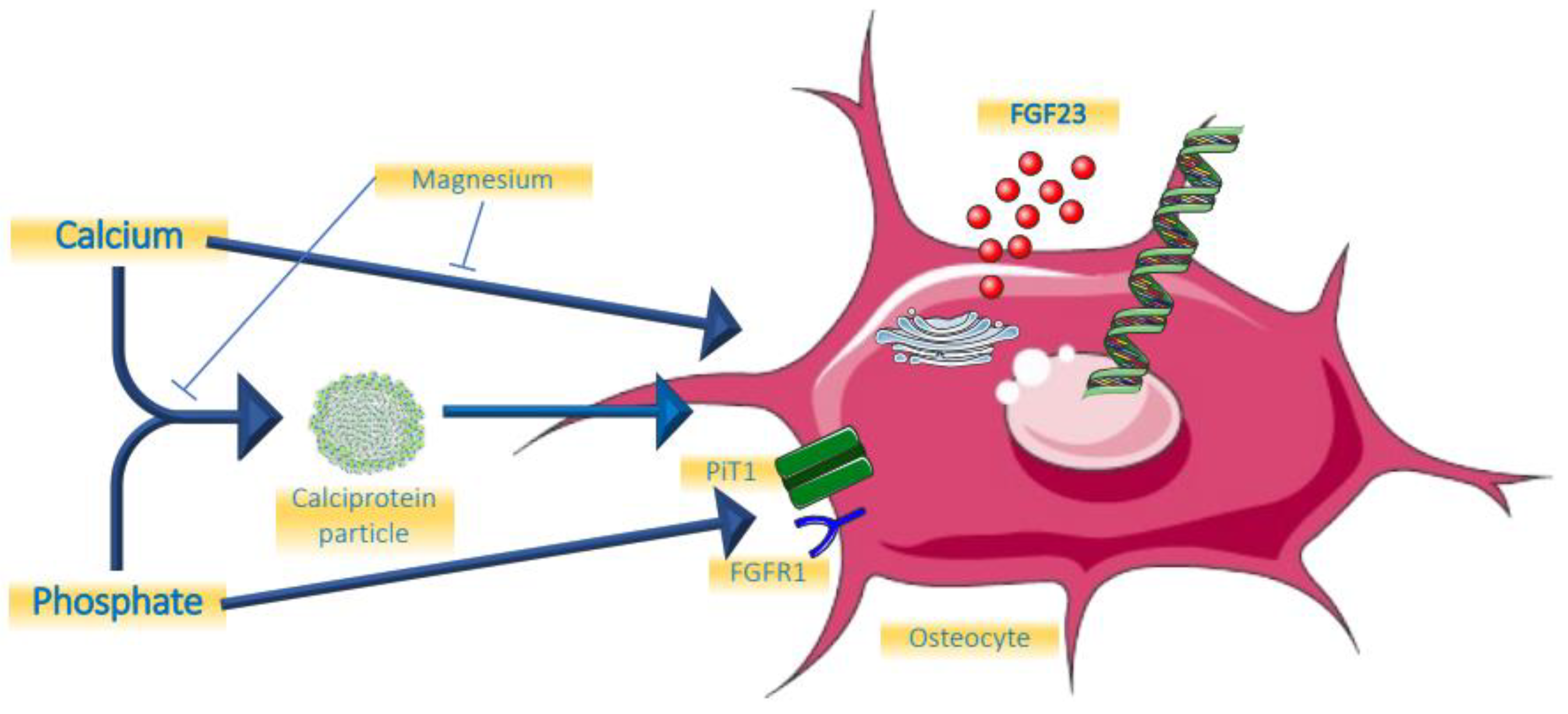
Figure 1. Effects of minerals on FGF23.
3. Hormonal Regulation of FGF23
3.1. Parathyroid Hormone
PTH was shown to be a relevant regulator of FGF23 by directly increasing its expression in bone in an experimental model of CKD [36]. Moreover, in that same study, parathyroidectomy before the onset of CKD completely abolished the FGF23 increment, even in a subsequent setting of hyperphosphatemia. This is probably mediated by the receptor PTH1R for PTH on bone cells, the same receptor that is involved in regulating bone turnover, and with Nuclear Receptor Related-1 protein (Nurr1) an intermediate intracellular molecule [37]. Another established action of PTH on bone cells is the suppression of the gene encoding for sclerostin (Sost). Sclerostin acts as local inhibitor of the Wnt pathway by sclerostin, thereby suppressing FGF23 [38][39][40]. PTH therefore unleashes FGF23 by suppressing sclerostin. Clinical studies suggest a biphasic response to PTH. In a short term (3 h) 1–34 PTH infusion in healthy young persons, FGF23 declined, most likely driven by PTH-induced renal phosphate loss [41]. During this period 1,25 dihydroxyvitamin D3 (1,25D) started to rise, which expectedly would induce increased dietary phosphate uptake. This, and the potential direct effects of PTH on bone cells may be the dominating effect following more prolonged exposure, giving rise to FGF23 increments. This indeed was suggested by a two days PTH infusion study that led to increased cFGF23 in healthy persons and people treated by dialysis regardless of bone turnover status [42]. Like for many other aspects, however, the role of PTH is complex, because if endogenous levels rise as a consequence of a decline of serum calcium by sodium citrate infusion, FGF23 did not increase [18]. Obviously, the stimulating effects of PTH on FGF23 may have been nullified by the low levels of calcium. There seems to be a logical physiological basis for the induction of FGF23 by PTH. The key purpose of PTH is to restore hypocalcemia and it does so in part by liberating calcium form bone. This is paralleled by release of phosphate, which is, besides by phosphaturic effects of PTH itself, excreted by the kidneys under the influence of FGF23.
Observations of persons with dialysis-dependent end-stage kidney disease treated with calcimimetics appear to be in line with the notion that lowering PTH is accompanied by declining FGF23 [43][44]. Remarkably, however, in both of these clinical studies, using the oral cinacalcet or the intravenous etelcalcetide, the decline of FGF23 followed reductions of phosphate and calcium, instead of PTH reductions.
3.2. Vitamin D
There is strong evidence that 1,25D directly induces Fgf23 gene transcription. Mice injected with the active form of vitamin D had increased levels of FGF23 mRNA, exclusively in bone, which was accompanied by a rise in serum FGF23 levels [45]. In that same study, rat-derived UMR-106 osteoblast-like cells had a 1000-fold increase of FGF23 mRNA 4 h after exposure to 1,25D. In another study with a focus on exploring the Fgf23 gene promotor region, this role of 1,25D was confirmed [46]. Collins and co-workers observed three patients that received a high dose of calcitriol after parathyroidectomy after surgery and observed steep increments of FGF23 [47]. Many clinical trials have been performed in which either active or nutritional vitamin D was the key intervention. In several of these trials, FGF23 levels were part of the follow-up parameters. The results of these observations have been summarized in two meta-analyses. In the first of these it was found that in patients that were deficient in vitamin D at baseline, the intervention induced a statistically significant increase of iFGF23 [48]. There was also an increase of cFGF23, but this did not reach statistical significance. A very recent meta-analysis could not confirm this effect of vitamin D, but in this meta-analysis, trials were included where participants did not have vitamin D deficiency at baseline, which may explain the discrepancy with the previous analysis [49]. In a study among children treated by dialysis, active vitamin D compounds (calcitriol or doxercalciferol) induced a substantial increase in FGF23 [50]. Collectively, these studies strongly suggest that vitamin D, especially active vitamin D, induces FGF23.
A summary of the roles of PTH and vitamin D is provided in Figure 2.
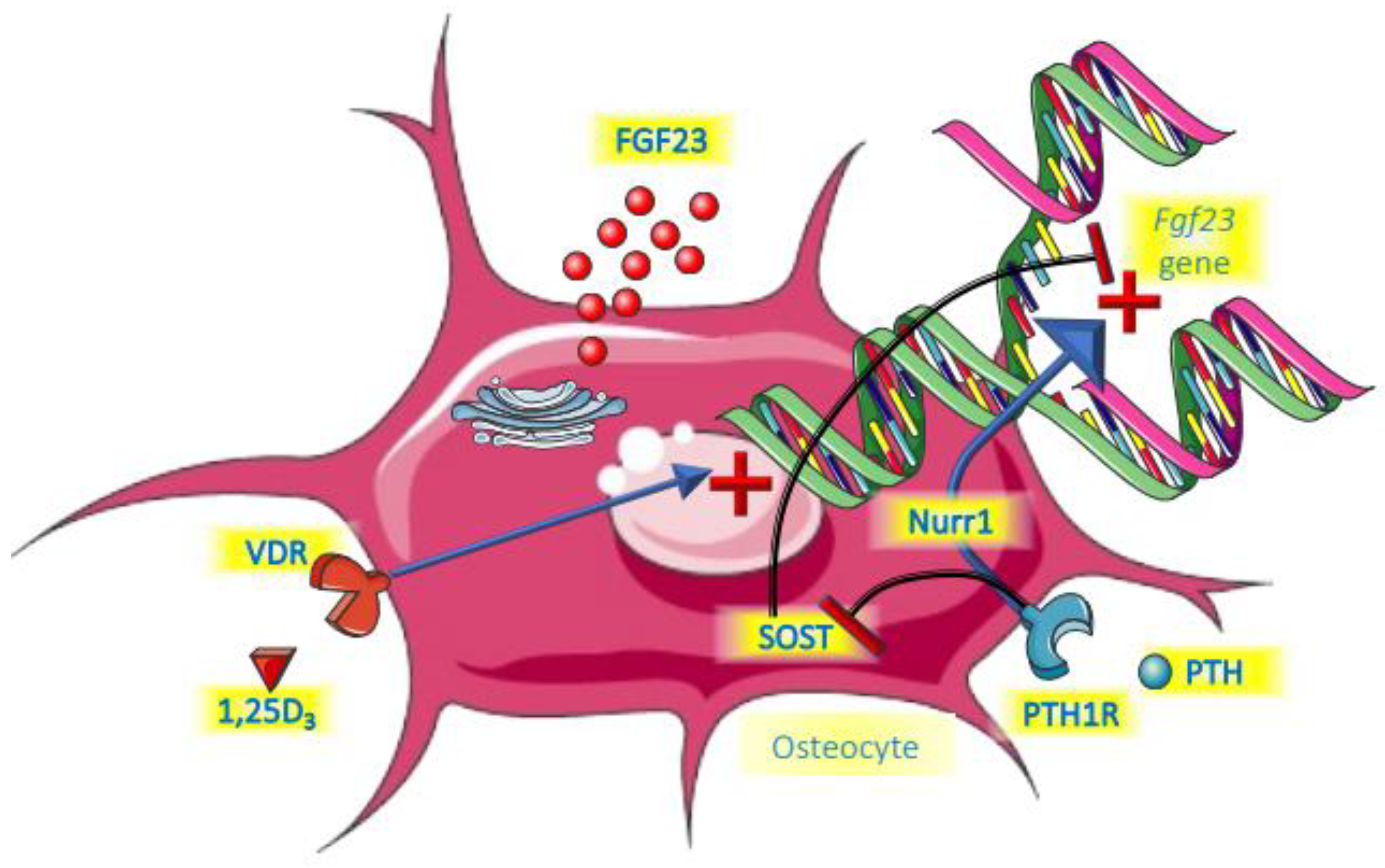
Figure 2. Endocrine control of FGF23.
References
- Vervloet, M.G.; van Ittersum, F.J.; Buttler, R.M.; Heijboer, A.C.; Blankenstein, M.A.; ter Wee, P.M. Effects of dietary phosphate and calcium intake on fibroblast growth factor-23. Clin. J. Am. Soc. Nephrol. 2011, 6, 383–389.
- Sigrist, M.; Tang, M.; Beaulieu, M.; Espino-Hernandez, G.; Er, L.; Djurdjev, O.; Levin, A. Responsiveness of FGF-23 and mineral metabolism to altered dietary phosphate intake in chronic kidney disease (CKD): Results of a randomized trial. Nephrol. Dial. Transplant. 2013, 28, 161–169.
- Ferrari, S.L.; Bonjour, J.P.; Rizzoli, R. Fibroblast growth factor-23 relationship to dietary phosphate and renal phosphate handling in healthy young men. J. Clin. Endocrinol. Metab. 2005, 90, 1519–1524.
- Tavana, N.; Thilakavathy, K.; Kennerson, M.L.; Ting, T.H. Genetic basis of hereditary hypophosphataemic rickets and phenotype presentation in children and adults. Endokrynol. Pol. 2021, 72, 366–394.
- Gattineni, J.; Baum, M. Genetic disorders of phosphate regulation. Pediatr. Nephrol. 2012, 27, 1477–1487.
- Bon, N.; Frangi, G.; Sourice, S.; Guicheux, J.; Beck-Cormier, S.; Beck, L. Phosphate-dependent FGF23 secretion is modulated by PiT2/Slc20a2. Mol. Metab. 2018, 11, 197–204.
- Takashi, Y.; Kosako, H.; Sawatsubashi, S.; Kinoshita, Y.; Ito, N.; Tsoumpra, M.K.; Nangaku, M.; Abe, M.; Matsuhisa, M.; Kato, S.; et al. Activation of unliganded FGF receptor by extracellular phosphate potentiates proteolytic protection of FGF23 by its O-glycosylation. Proc. Natl. Acad. Sci. USA 2019, 116, 11418–11427.
- Hori, M.; Kinoshita, Y.; Taguchi, M.; Fukumoto, S. Phosphate enhances Fgf23 expression through reactive oxygen species in UMR-106 cells. J. Bone Miner. Metab. 2016, 34, 132–139.
- Tsai, W.-C.; Wu, H.-Y.; Peng, Y.-S.; Hsu, S.-P.; Chiu, Y.-L.; Chen, H.-Y.; Yang, J.-Y.; Ko, M.-J.; Pai, M.-F.; Tu, Y.-K.; et al. Effects of lower versus higher phosphate diets on fibroblast growth factor-23 levels in patients with chronic kidney disease: A systematic review and meta-analysis. Nephrol. Dial. Transplant. 2018, 33, 1977–1983.
- Bouma-de Krijger, A.; van Ittersum, F.J.; Hoekstra, T.; Ter Wee, P.M.; Vervloet, M.G. Short-term effects of sevelamer-carbonate on fibroblast growth factor 23 and pulse wave velocity in patients with normophosphataemic chronic kidney disease Stage 3. Clin. Kidney J. 2019, 12, 678–685.
- Kovesdy, C.P.; Lu, J.L.; Wall, B.M.; Gyamlani, G.; Naseer, A.; Wallick, A.; Han, Z.; Thomas, F.; Quarles, L.D.; Jarmukli, N. Changes With Lanthanum Carbonate, Calcium Acetate, and Phosphorus Restriction in CKD: A Randomized Controlled Trial. Kidney Int. Rep. 2018, 3, 897–904.
- Ketteler, M.; Sprague, S.M.; Covic, A.C.; Rastogi, A.; Spinowitz, B.; Rakov, V.; Walpen, S.; Floege, J. Effects of sucroferric oxyhydroxide and sevelamer carbonate on chronic kidney disease-mineral bone disorder parameters in dialysis patients. Nephrol. Dial. Transplant. 2018, 34, 1163–1170.
- Soriano, S.; Ojeda, R.; Rodríguez, M.; Almadén, Y.; Rodriguez, M.; Martín-Malo, A.; Aljama, P. The effect of phosphate binders, calcium and lanthanum carbonate on FGF23 levels in chronic kidney disease patients. Clin. Nephrol. 2013, 80, 17–22.
- Rodriguez-Ortiz, M.E.; Lopez, I.; Muñoz-Castañeda, J.R.; Martínez, J.; Peralta-Ramírez, A.; Pineda, C.; Canalejo, A.; Jaeger, P.; Aguilera-Tejero, E.; Rodriguez, M.; et al. Calcium deficiency reduces circulating levels of FGF23. J. Am. Soc. Nephrol. 2012, 23, 1190–1197.
- David, V.; Dai, B.; Martin, A.; Huang, J.; Han, X.; Quarles, L.D. Calcium regulates FGF-23 expression in bone. Endocrinology 2013, 154, 4469–4482.
- Shikida, Y.; Mizobuchi, M.; Inoue, T.; Hamada, T.; Ogata, H.; Koiwa, F.; Shibata, T. Effect of Continuous Intravenous Calcium Loading on Fibroblast Growth Factor 23 in Normal and Uremic Rats. Calcif. Tissue Int. 2018, 103, 455–464.
- Han, N.; Hong, S.H.; Kim, Y.S.; Kim, D.K.; Kim, I.-W.; Ji, E.; Oh, J.M. Effect of additive calcium administration on FGF23 levels in patients with mild chronic kidney disease treated with calcitriol: A randomized, open-labeled clinical trial. Ther. Clin. Risk Manag. 2017, 13, 999–1007.
- Wesseling-Perry, K.; Wang, H.; Elashoff, R.; Gales, B.; Jüppner, H.; Salusky, I.B. Lack of FGF23 response to acute changes in serum calcium and PTH in humans. J. Clin. Endocrinol. Metab. 2014, 99, E1951–E1956.
- Heiss, A.; DuChesne, A.; Denecke, B.; Grötzinger, J.; Yamamoto, K.; Renné, T.; Jahnen-Dechent, W. Structural basis of calcification inhibition by alpha 2-HS glycoprotein/fetuin-A. Formation of colloidal calciprotein particles. J. Biol. Chem. 2003, 278, 13333–13341.
- Heiss, A.; Pipich, V.; Jahnen-Dechent, W.; Schwahn, D. Fetuin-A is a mineral carrier protein: Small angle neutron scattering provides new insight on Fetuin-A controlled calcification inhibition. Biophys. J. 2010, 99, 3986–3995.
- Holt, S.G.; Smith, E.R. Fetuin-A-containing calciprotein particles in mineral trafficking and vascular disease. Nephrol. Dial. Transplant. 2016, 31, 1583–1587.
- Gangneux, C.; Daveau, M.; Hiron, M.; Derambure, C.; Papaconstantinou, J.; Salier, J.P. The inflammation-induced down-regulation of plasma Fetuin-A (alpha2HS-Glycoprotein) in liver results from the loss of interaction between long C/EBP isoforms at two neighbouring binding sites. Nucleic Acids Res. 2003, 31, 5957–5970.
- Mutluay, R.; Değertekin, C.K.; Sayilar, E.I.; Derici, Ü.; Gültekin, S.; Gönen, S.; Arinsoy, S.T. Serum fetuin-A is associated with the components of MIAC(malnutrition, inflammation, atherosclerosis, calcification) syndrome in different stages of chronic kidney disease. Turk. J. Med. Sci. 2019, 49, 327–335.
- Cozzolino, M.; Galassi, A.; Biondi, M.L.; Turri, O.; Papagni, S.; Mongelli, N.; Civita, L.; Gallieni, M.; Brancaccio, D. Serum fetuin-A levels link inflammation and cardiovascular calcification in hemodialysis patients. Am. J. Nephrol. 2006, 26, 423–429.
- Iwazu, Y.; Kuro, O.M.; Miura, Y.; Takeda, S.I.; Yamada, T.; Nagata, D. Calciprotein particles and fibroblast growth factor 23 contribute to the pathophysiology of hypercalcemia in a patient with renal sarcoidosis. Clin. Kidney J. 2021, 14, 421–423.
- Nakamura, K.; Nagata, Y.; Hiroyoshi, T.; Isoyama, N.; Fujikawa, K.; Miura, Y.; Matsuyama, H.; Kuro-O, M. The effect of lanthanum carbonate on calciprotein particles in hemodialysis patients. Clin. Exp. Nephrol. 2020, 24, 323–329.
- Miura, Y.; Iwazu, Y.; Shiizaki, K.; Akimoto, T.; Kotani, K.; Kurabayashi, M.; Kurosu, H.; Kuro-O, M. Identification and quantification of plasma calciprotein particles with distinct physical properties in patients with chronic kidney disease. Sci. Rep. 2018, 8, 1256.
- Akiyama, K.-I.; Miura, Y.; Hayashi, H.; Sakata, A.; Matsumura, Y.; Kojima, M.; Tsuchiya, K.; Nitta, K.; Shiizaki, K.; Kurosu, H.; et al. Calciprotein particles regulate fibroblast growth factor-23 expression in osteoblasts. Kidney Int. 2020, 97, 702–712.
- Bressendorff, I.; Hansen, D.; Schou, M.; Pasch, A.; Brandi, L. The Effect of Increasing Dialysate Magnesium on Serum Calcification Propensity in Subjects with End Stage Kidney Disease: A Randomized, Controlled Clinical Trial. Clin. J. Am. Soc. Nephrol. 2018, 13, 1373–1380.
- Pasch, A.; Farese, S.; Gräber, S.; Wald, J.; Richtering, W.; Floege, J.; Jahnen-Dechent, W. Nanoparticle-based test measures overall propensity for calcification in serum. J. Am. Soc. Nephrol. 2012, 23, 1744–1752.
- Moor, M.B.; Ramakrishnan, S.K.; Legrand, F.; Bachtler, M.; Koesters, R.; Hynes, N.E.; Pasch, A.; Bonny, O. Elevated serum magnesium lowers calcification propensity in Memo1-deficient mice. PLoS ONE 2020, 15, e0236361.
- van den Broek, D.H.N.; Chang, Y.M.; Elliott, J.; Jepson, R.E. Prognostic importance of plasma total magnesium in a cohort of cats with azotemic chronic kidney disease. J. Vet. Intern. Med. 2018, 32, 1359–1371.
- Kosk, D.; Kramer, H.; Luke, A.; Camacho, P.; Bovet, P.; Rhule, J.P.; Forrester, T.; Wolf, M.; Sempos, C.; Melamed, M.L.; et al. Dietary factors and fibroblast growth factor-23 levels in young adults with African ancestry. J. Bone Miner. Metab. 2017, 35, 666–674.
- Matsuzaki, H.; Katsumata, S.; Maeda, Y.; Kajita, Y. Changes in circulating levels of fibroblast growth factor 23 induced by short-term dietary magnesium deficiency in rats. Magnes. Res. 2016, 29, 48–54.
- Leenders, N.H.J.; Vervloet, M.G. Magnesium: A Magic Bullet for Cardiovascular Disease in Chronic Kidney Disease? Nutrients 2019, 11, 455.
- Lavi-Moshayoff, V.; Wasserman, G.; Meir, T.; Silver, J.; Naveh-Many, T. PTH increases FGF23 gene expression and mediates the high-FGF23 levels of experimental kidney failure: A bone parathyroid feedback loop. Am. J. Physiol. Ren. Physiol. 2010, 299, F882–F889.
- Meir, T.; Durlacher, K.; Pan, Z.; Amir, G.; Richards, W.G.; Silver, J.; Naveh-Many, T. Parathyroid hormone activates the orphan nuclear receptor Nurr1 to induce FGF23 transcription. Kidney Int. 2014, 86, 1106–1115.
- Wein, M.N.; Liang, Y.; Göransson, O.; Sundberg, T.B.; Wang, J.; Williams, E.A.; O’Meara, M.J.; Govea, N.; Beqo, B.; Nishimori, S.; et al. SIKs control osteocyte responses to parathyroid hormone. Nat. Commun. 2016, 7, 13176.
- Nagata, Y.; Imanishi, Y.; Tateishi, T.; Miyaoka, D.; Kurajoh, M.; Arnold, A.; Emoto, M. Parathyroid Hormone Regulates Circulating Levels of Sclerostin and FGF23 in a Primary Hyperparathyroidism Model. J. Endocr. Soc. 2022, 6, bvac027.
- Carpenter, K.A.; Davison, R.; Shakthivel, S.; Anderson, K.D.; Ko, F.C.; Ross, R.D. Sclerostin antibody improves phosphate metabolism hormones, bone formation rates, and bone mass in adult Hyp mice. Bone 2022, 154, 116201.
- Gutierrez, O.M.; Smith, K.T.; Barchi-Chung, A.; Patel, N.M.; Isakova, T.; Wolf, M. (1-34) Parathyroid hormone infusion acutely lowers fibroblast growth factor 23 concentrations in adult volunteers. Clin. J. Am. Soc. Nephrol. 2012, 7, 139–145.
- Wesseling-Perry, K.; Harkins, G.C.; Wang, H.-J.; Elashoff, R.; Gales, B.; Horwitz, M.J.; Stewart, A.F.; Jüppner, H.; Salusky, I.B. The calcemic response to continuous parathyroid hormone (PTH)(1-34) infusion in end-stage kidney disease varies according to bone turnover: A potential role for PTH(7-84). J. Clin. Endocrinol. Metab. 2010, 95, 2772–2780.
- Koizumi, M.; Komaba, H.; Nakanishi, S.; Fujimori, A.; Fukagawa, M. Cinacalcet treatment and serum FGF23 levels in haemodialysis patients with secondary hyperparathyroidism. Nephrol. Dial. Transplant. 2012, 27, 784–790.
- Wolf, M.; Block, G.A.; Chertow, G.M.; Cooper, K.; Fouqueray, B.; Moe, S.M.; Sun, Y.; Tomlin, H.; Vervloet, M.; Oberbauer, R. Effects of etelcalcetide on fibroblast growth factor 23 in patients with secondary hyperparathyroidism receiving hemodialysis. Clin. Kidney J. 2020, 13, 75–84.
- Kolek, O.I.; Hines, E.R.; Jones, M.D.; LeSueur, L.K.; Lipko, M.A.; Kiela, P.R.; Collins, J.F.; Haussler, M.R.; Ghishan, F.K. 1alpha,25-Dihydroxyvitamin D3 upregulates FGF23 gene expression in bone: The final link in a renal-gastrointestinal-skeletal axis that controls phosphate transport. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G1036–G1042.
- Ito, M.; Sakai, Y.; Furumoto, M.; Segawa, H.; Haito, S.; Yamanaka, S.; Nakamura, R.; Kuwahata, M.; Miyamoto, K.-I. Vitamin D and phosphate regulate fibroblast growth factor-23 in K-562 cells. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E1101–E1109.
- Collins, M.T.; Lindsay, J.R.; Jain, A.; Kelly, M.H.; Cutler, C.M.; Weinstein, L.S.; Liu, J.; Fedarko, N.; Winer, K.K. Fibroblast growth factor-23 is regulated by 1alpha,25-dihydroxyvitamin D. J. Bone Miner. Res. 2005, 20, 1944–1950.
- Charoenngam, N.; Rujirachun, P.; Holick, M.F.; Ungprasert, P. Oral vitamin D3 supplementation increases serum fibroblast growth factor 23 concentration in vitamin D-deficient patients: A systematic review and meta-analysis. Osteoporos. Int. 2019, 30, 2183–2193.
- Meshkini, F.; Soltani, S.; Clark, C.C.; Tam, V.; Meyre, D.; Toupchian, O.; Saraf-Bank, S.; Abdollahi, S. The effect of vitamin D supplementation on serum levels of fibroblast growth factor- 23: A systematic review and meta-analysis of randomized controlled trials. J. Steroid Biochem. Mol. Biol. 2022, 215, 106012.
- Wesseling-Perry, K.; Pereira, R.C.; Sahney, S.; Gales, B.; Wang, H.-J.; Elashoff, R.; Jüppner, H.; Salusky, I.B. Calcitriol and doxercalciferol are equivalent in controlling bone turnover, suppressing parathyroid hormone, and increasing fibroblast growth factor-23 in secondary hyperparathyroidism. Kidney Int. 2011, 79, 112–119.
More
Information
Subjects:
Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
786
Revisions:
2 times
(View History)
Update Date:
17 May 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No