Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Marialucia Milite | -- | 3341 | 2022-05-12 17:43:39 | | | |
| 2 | Conner Chen | Meta information modification | 3341 | 2022-05-13 02:56:38 | | | | |
| 3 | Conner Chen | Meta information modification | 3341 | 2022-05-13 02:59:10 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Milite, M.; Prandi, F.R.; , .; Celotto, R.; Lerakis, S.; Romeo, F. Pathophysiology of Coronary Artery Disease in Diabetes Mellitus. Encyclopedia. Available online: https://encyclopedia.pub/entry/22884 (accessed on 23 July 2026).
Milite M, Prandi FR, , Celotto R, Lerakis S, Romeo F. Pathophysiology of Coronary Artery Disease in Diabetes Mellitus. Encyclopedia. Available at: https://encyclopedia.pub/entry/22884. Accessed July 23, 2026.
Milite, Marialucia, Francesca Romana Prandi, , Roberto Celotto, Stamatios Lerakis, Francesco Romeo. "Pathophysiology of Coronary Artery Disease in Diabetes Mellitus" Encyclopedia, https://encyclopedia.pub/entry/22884 (accessed July 23, 2026).
Milite, M., Prandi, F.R., , ., Celotto, R., Lerakis, S., & Romeo, F. (2022, May 12). Pathophysiology of Coronary Artery Disease in Diabetes Mellitus. In Encyclopedia. https://encyclopedia.pub/entry/22884
Milite, Marialucia, et al. "Pathophysiology of Coronary Artery Disease in Diabetes Mellitus." Encyclopedia. Web. 12 May, 2022.
Copy Citation
Diabetes mellitus (DM) significantly increases the risk of cardiovascular diseases (CVD), all-cause mortality and cardiovascular mortality. According to the Framingham study, patients with DM have a two-fold to four-fold increased risk of developing coronary artery disease (CAD) and myocardial infarction (MI) and a four-fold to six-fold increased risk of developing congestive heart failure (HF). Hyperglycemia represents the main initiating factor in the pathogenesis of diabetic complications.
diabetes mellitus
hyperglycemia
coronary artery disease
1. Pathophysiology of CAD in Diabetes Mellitus
Diabetes is a complex metabolic process that induces atherosclerosis development and/or accelerates its progression through a multifactorial process. Vascular complications associated with hyperglycemia in diabetes often begin with endothelial dysfunction; however, the mechanisms underlying the pathogenesis of CAD in diabetic patients are still not completely understood [1]. Most of the reversible risk factors for atherosclerosis are more prevalent among diabetic patients, including hypertension, obesity, dyslipidemia, hyperglycemia and hyperinsulinemia [2].
However, the contribution of the higher prevalence of all these classic risk factors in terms explains only a portion (25%) of the excess of coronary heart disease risk in T2DM [3]. This discrepancy is in part related to changes in the traditionally measured risk factors that occur in diabetes, including glycation and advanced glycation end products (AGEs), glycoxidation and oxidation, insulin resistance and hyperinsulinemia, abnormalities of apoprotein and lipoprotein particle distribution and procoagulant states [2]. Other factors to be considered are the possible adverse effects of hypoglycemic agents, the influence of diabetic cardiomyopathy, inflammation, genetic and epigenetic modifications.
Inflammation plays an important role in the pathophysiological link between DM and atherosclerosis. Vircow first recognized the role of inflammation in the pathogenesis of atherosclerosis [4]. Chronic inflammation and inflammasome activation play important roles in the pathogenesis of T2DM. Patients with T2DM have significantly increased messenger RNA (mRNA) and protein levels of the NLRP3 inflammasome and increased proinflammatory cytokines in monocyte-derived macrophages [5]. Patients with DM present more intense pro-inflammatory, pro-oxidant and pro-thrombotic stimuli; the latter is related to hyper-reactive platelets, the upregulation of pro-thrombotic markers and suppression of fibrinolysis [6].
Jarrett in 1984 [7] and Stern, in 1995 [8], suggested that DM and atherosclerosis have a common genetic and environmental background, rather than one being a complication of the other (the “common soil” hypothesis). Insulin-resistance and hyperglycemia, inflammation, oxidative stress, hypercoagulability, high blood pressure, dyslipidemia and obesity are common pathophysiological factors in T2DM and CVD. Many common single-nucleotide polymorphisms (SNP) have been associated with an increased risk of CVD and T2DM [9].
Genome-wide association studies have been employed to search for gene predispositions to both diseases. The main genes whose variants are commonly associated with both T2DM and CVD are involved in LDL oxidation (paraxonase, polymorphism Gln-Arg 192 [10] or Met-Leu 54 [11], which lead to decreased levels or activity of paraxonase, an enzyme that normally protects LDL from proatherogenic modifications), redox balance (superoxide dismutase 2 and polymorphism Ala-Val 16, which lead to decreased SOD2 activity) [12], the adiponectin pathway (adiponectin, polymorphism +276 G/T [13] and adiponectin receptor ADIPOR1, haplotypes rs7539542, rs10920531 and rs4950894 [14].
These polymorphisms determine the reduced activity of the adiponectin pathway that normally has anti-inflammatory and antiatherogenic effects), lipoprotein transport (ApoE, polymorphism Arg112/Arg158 that encodes apoE4 isoform, which has a higher affinity for LDL-R, leading to early receptor occupation, accumulation of LDL particles, LDL-R synthesis suppression and lower clearance of lipoproteins from the body through LDL-R) [15] but also cell cycle regulation (CDKN2A/2B, variant rs4977574) [16], cell growth, differentiation and glucose metabolism (HMGA1, variant rs146052672, which is associated with increased susceptibility to T2DM and acute MI) [17].
A proteomic analysis of a T2DM population in relation to coronary phenotyping highlighted the potential role of GDF15, renin, adiponectin, serine protease HTRA1 and tetranectin in the development of T2DM and CAD [18]. There are also some genes with polymorphisms that are associated with a reduction of risk for CVD and T2DM, such as genes involved in the cholesterol metabolism (PCSK9), cells mitosis (PSRC1) or intracellular trafficking (SORT1) [9]. In addition to biochemical mechanisms and genetic factors, epigenetic mechanisms play a key role in the physiopathology of DM-induced CAD.
2. Role of Lipid and Glucose Metabolism in the Pathophysiological Link between Diabetes and Atherosclerosis
DM is a glucose metabolism disorder characterized by chronic hyperglycemia resulting from a deficit of insulin production and/or action. Both T1DM and T2DM cause hyperglycemia, which leads to endothelial dysfunction through different pathways. In addition, T2DM also causes insulin resistance, which is another determinant for endothelial dysfunction. Obesity, which is an independent risk factor for endothelial cell dysfunction is also closely related to type 2 diabetes [19].
Hyperglycemia represents the main initiating factor in the pathogenesis of diabetic complications [20] (Figure 1). A causal relationship between levels of hyperglycemia and diabetic microvascular and macrovascular complications was found in both T1DM and T2DM patients [21]. Intensive diabetes therapy and lower levels of HbA1c in T1DM are associated with thinner carotid IMT, less coronary calcification and a lower incidence of clinical cardiovascular events, such as myocardial infarction, stroke and cardiac death [22]. In T2DM patients, metabolic control (HbA1c < 7%) reduces the risk of coronary heart disease death and all coronary heart disease events [23]. Glycemic control is therefore of fundamental importance to reduce the progression of diabetes and its microvascular and macrovascular complications.
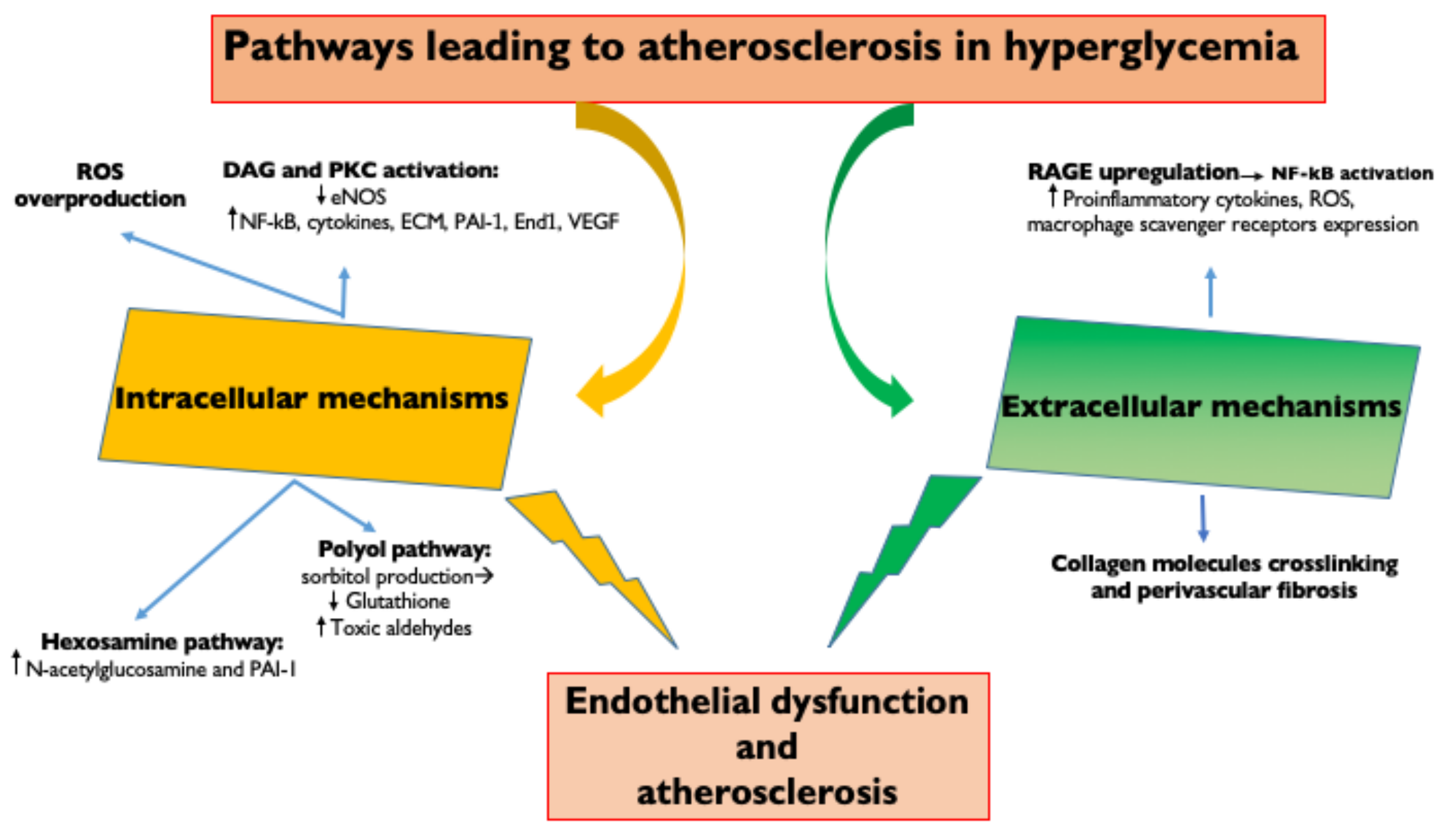
Figure 1. Pathways leading to atherosclerosis in hyperglycemia. Intracellular and extracellular molecular mechanisms underlying hyperglycemia-induced endothelial dysfunction. ROS, Reactive oxygen species; DAG, diacylglycerol; PKC, Protein kinase C; eNOS, endothelial Nitric Oxide Synthase; NFkB, Nuclear Factor κB; ECM, extracellular matrix; PAI—1, Plasminogen activator inhibitor-1; End1, endothelin 1; VEGF, Vascular endothelial growth factor; AGE, Advanced glycation end products; and RAGE, receptor for advanced glycation end products.
Vascular endothelial cells are a major target of hyperglycemic damage; however, the mechanisms underlying this damage are still incompletely understood. Hyperglycemia is linked to atherosclerosis through both extracellular and intracellular mechanisms.
The extracellular mechanisms are based on the fact that chronic hyperglycemia induces the non-enzymatic glycation of proteins, lipids and lipoproteins with the generation of AGEs [24]. AGEs upregulate their receptors (RAGE) leading to the activation of transcription factors, such as nuclear factor-κB (NF-κB) and its target genes [24]. This results in the production of pro-inflammatory cytokines (IL-1β, IL-6, IL-18 and TNFα) [25], generation of ROS, endothelial expression of adhesion molecules (vascular cell adhesion molecule 1 (VCAM-1) and intercellular cell adhesion molecule 1 (ICAM-1), which promote monocyte entry into the subendothelium), increased production of endothelin-1 and reduced generation of nitric oxide (that enhance vasoconstriction) [1] and increased macrophage expression of scavenger receptors (SR: CD36 and SR class A1, which promote macrophage phagocytosis) [26].
AGE formation on vascular cells also determines the cross-linking of collagen molecules with an increased perivascular fibrosis, which leads to a reduction in arterial compliance and also to coronary microvascular stenosis and microaneurysms [27]. LDL glycation increases the atherogenic potential of LDL: small dense LDL particles (LDL molecules desialylated) glycation favors interactions with subendothelial proteoglycans, increasing the LDL retention time in the subendothelial space and LDL phagocytosis by macrophages to form foam cells [28].
Small, dense LDL are also more atherogenic because of their lower binding affinity to LDL receptors and lower resistance to oxidative stress [1]. High intracellular glucose levels activate metabolic pathways that result in reactive oxygen species (ROS) overproduction, the activation of PKC, the hexosamine pathway and the polyol pathway. The increased inflammatory response leads to endothelial dysfunction with augmented vascular permeability and impaired vasodilation as well as an associated increased risk of leukocyte/platelet adhesion, thrombosis and inflammation [26].
Hyperglycemia determines directly (through an increase in diacylglycerol) or indirectly (through the oxidative stress and the increase of ROS) increased protein-kinase C (PKC) signaling, which can upregulate NF-κB and downregulate eNOS [29], stimulate the production of cytokines, the extracellular matrix, the fibrinolytic inhibitor plasminogen activator inhibitor (PAI-1), the vasoconstrictor endothelin-1 and VEGF. These changes lead to endothelial dysfunction due to the increase of vascular permeability, basement membrane thickening, vascular occlusion and angiogenesis [20].
High intracellular glucose also results in increased hexosamine pathway flux, which leads to increased N-acetylglucosamine and upregulation of PAI-1 [30]. Finally, when intracellular glucose levels are high, there is increased polyol pathway flux. A part of the glucose is reduced to sorbitol by aldose reductase, which competes for the cofactor NADPH with glutathione reductase. This deprives the cell of reduced glutathione, an important antioxidant, with the consequent ineffective reduction of toxic aldehydes, which accumulate in the cell [30].
IR is another important factor in the pathogenesis of T2DM complications and coronary vessel damage. The metabolic consequences of IR are hyperglycemia, impaired insulin secretion, oxidative stress, hypertension, dyslipidemia and increased visceral fat deposition. The pancreas attempts to overcome insulin resistance by increasing insulin secretion. Compensatory hyperinsulinemia determines exaggerated responses in the tissues that remain sensitive to insulin, with activation of the sympathetic nervous system and the increased reabsorption of renal sodium, leading to hypertension [31].
Insulin resistance in fat cells leads to increased lipolysis, fatty acid release, dyslipidemia and vascular abnormalities. Visceral fat is more resistant than subcutaneous fat to the action of insulin, and the release of fatty acids from visceral fat can directly (through PKC activation) block insulin-signaling pathways, thereby, increasing insulin resistance. Obesity, especially visceral obesity, is negatively correlated with insulin sensitivity [32].
3. Epigenetic Modifications
Epigenetic modifications are changes in gene function that are mitotically and/or meiotically heritable and that do not entail a change in DNA sequence [33]. They include DNA methylation, histone modifications and non-coding RNA (ncRNA)-mediated control (Figure 2). They are the result of interactions between environmental stimuli and the regulation mechanisms of DNA expression, representing a molecular link between environmental factors and complex diseases, including atherosclerosis and diabetes.
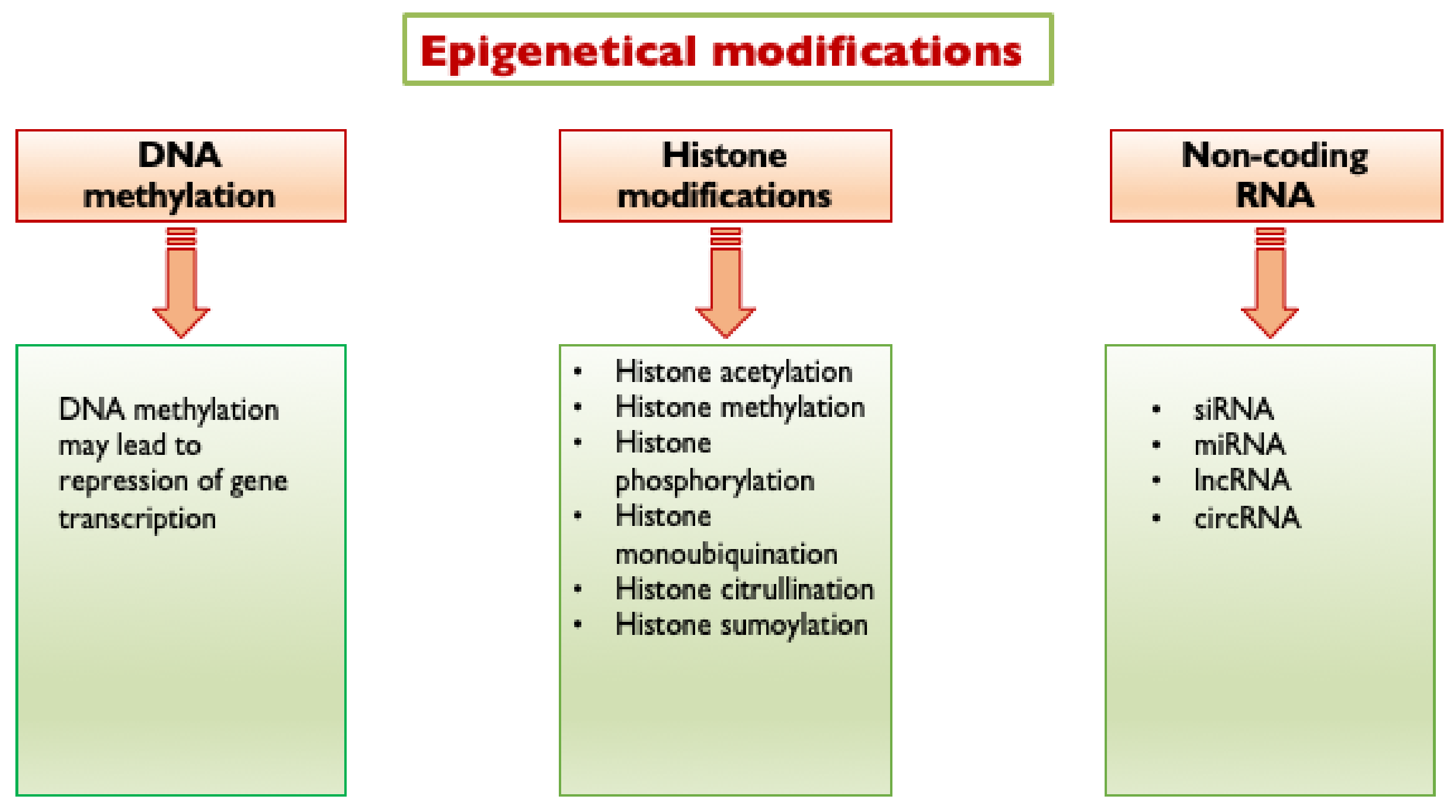
Figure 2. Epigenetic modifications triggered by hyperglycemia. Hyperglycemia triggers the following epigenetic modifications: DNA methylation, histone modifications and non-coding RNA mediated control. siRNA, short interfering RNA; miRNA, micro RNA; lncRNA, long non-coding RNA; and circRNA, circular RNA.
Epigenetic changes at DNA-histone complexes significantly alter gene transcription by modulating the chromatin accessibility. DNA methylation involves the addition of a methyl group to cytosines within cytosine/guanine pairs (CpG). Several types of histone modifications are known, including histone methylation, acetylation, phosphorylation, ubiquitination, SUMOylation (which is the attachment of a SUMO (Small Ubiquitin-like Modifier) peptide) and citrullination are the best studied and important in regulating chromatin structure and transcriptional activity.
DNA methylation within a gene promoter typically represses gene transcription. Histone acetylation generally determines increased transcription, while histone methylation has either a transcriptionally permissive or repressive character, depending on the location of the targeted amino acid residues and on the number of methyl groups added [34]. In detail, the methylation of H3K9 and H3K27 is transcriptionally repressive, while the methylation of H3K4 and H3K36 is transcriptionally permissive [35].
Histone phosphorylation and histone mono-ubiquitination can regulate transcriptional activity working in conjunction (“cross talk”) with other histone modifications, such as histone acetylation [34]. SUMO peptides can use the same lysine residues as ubiquitin; therefore, the SUMOylation of substrate proteins may generate proteins resistant to degradation by the ubiquitin proteasome system [36].
Histone citrullination is involved in the regulation of chromatin structure and gene transcriptional activity and engages in cross-talk with histones marked with other modifications [37]. Epigenetic modifications are performed by several specific enzymes, which can be classified as “writers”, and these modify discrete residues on histone tails adding marks that designate certain regions for transcriptional regulation (such as DNA methyltransferases and histone acetyltransferases) and “erasers”, which remove these marks (such as histone deacetylases and histone demethylases) [35].
DNA methylation is catalyzed by DNA methyltransferases (DNMT) [38], whereas ten-eleven translocation (TET) enzymes are responsible for DNA demethylation [35]. Histone acetylation is regulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs, further subdivided in four classes; those in class III are called sirtuins or SIRTs), while histone methylation is mediated by histone methyltransferases (HMTs) and histone demethylases (HDMs). Histone phosphorylation is controlled by kinases and phosphatases. Histone ubiquitination is conducted by histone ubiquitin ligases and histone deubiquitinating enzymes (DUBs) [34], while SUMOylation is by SUMO ligases and peptidases. Histone citrullination is mediated by peptidyl arginine deiminase (PAD) enzymes.
Epigenetic modifications are reversible, to ensure that specific genes can be expressed or silenced by specific stimulators. Shear stress, hyperglycemia, hyperlipidemia and aging are the most important factors that determinate epigenetic modifications for endothelial cells, vascular smooth muscle cell and circulating monocytes. Moreover, the epigenetic status is influenced by ncRNA molecules that regulate post-transcriptional modifications and control the mechanisms of translational inhibition [38].
4. Hyperglycemia-Induced Epigenetic Modifications and the Impact on Atherosclerosis
A growing amount of evidence suggest that, in addition to biochemical processes, hyperglycemia can also induce epigenetic modifications that lead to increased oxidative stress, PKC signaling, AGE formation, NFkB-dependent monocyte-chemotactic protein 1 (MCP-1) and VCAM1 expression, with consequent endothelial dysfunction and atherosclerosis [26].
Hyperglycemia determines DNA demethylation in endothelial cells through an upregulation of TETs. Endothelial cells exposed to transient hyperglycemia (7 days) are characterized by the persistence of NFκB-p65 gene (RELA) activation, induced by DNA demethylation of the NFκB promoter at CpGs islands through TET2 upregulation [39]. Hyperglycemia activates the NFκB-p65 gene in endothelial cells also by mono-methylation of lysine 4 on histone 3 (H3K4m1) through histone methyltransferase Set7 and demethylation of H3K9 on the p65 promoter by lysine-specific demethylase 1 [40].
Hyperglycemia also influences the SUMOylation of I𝜅B𝛼, which is the main inhibitor of NF-κB dimers: when IκBα is SUMOylated, it is resistant to ubiquitin-induced degradation, and it inhibits NF-κB. In high glucose conditions, the SUMOylation of I𝜅B𝛼 is decreased, with consequent activation of the NF-κB pathway [41]. The NFκB upregulation leads to increasing its gene targets, including adhesion molecules, cytokines and chemokines (such as VCAM1, IL-6, TNFα and MCP-1), causing vascular inflammation and atherosclerosis (Figure 3). Ubiquitination and SUMOylation can also activate TGF-β and MAPK and inhibit Nrf2-oxidative stress [36]. Ubiquitin-proteasome system overactivity, by contributing to the increased inflammation process, may enhance the risk of complication during myocardial ischemia in diabetic patients [42].
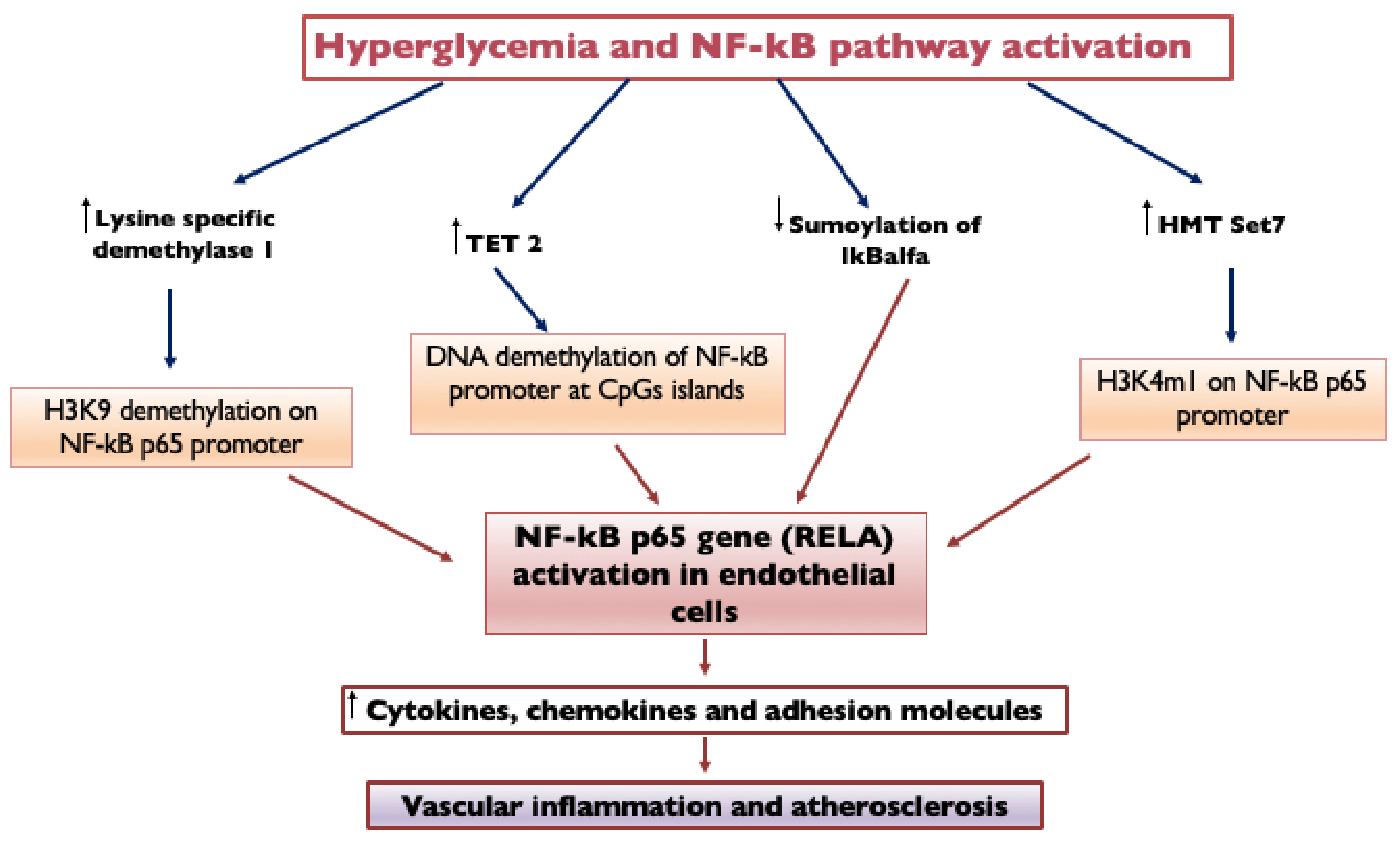
Figure 3. Hyperglycemia and NF-kB pathway activation. Endothelial cells exposed to transient hyperglycemia are characterized by the persistence of NFκB-p65 gene (RELA) activation, induced by DNA demethylation of the NF-κB promoter at CpGs islands through TET2 upregulation. Hyperglycemia activates the NF-κB-p65 gene in endothelial cells also by mono-methylation of lysine 4 on histone 3 (H3K4m1) through histone methyltransferase Set7 and demethylation of H3K9 on the p65 promoter by lysine-specific demethylase 1. Hyperglycemia also influences the SUMOylation of I𝜅B𝛼, which is the main inhibitor of NF-κB dimers: when IκBα is SUMOylated, it is resistant to ubiquitin-induced degradation, and it inhibits NF-κB; in high glucose conditions, SUMOylation of I𝜅B𝛼 is decreased, with consequent activation of the NF-κB pathway. NF-κB upregulation leads to increasing its gene targets, causing vascular inflammation and atherosclerosis. NF-kB, Nuclear Factor κB; TET2, Ten-eleven translocation 2; IκBα, nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor alpha; HMT, Histone lysine methyltransferase; H3K9 demethylation, Demethylation of Histone H3 at Lysine 9; and H3K4m1, Monomethylation on lysine 4 of histone H3.
The activation process of Set7 takes also place in circulating monocytes, determining the methylation of H3K4m1 on the NFκB-p65 promoter, with activation of the transcription of genes responsible for the inflammatory process and for the increased adhesion capacity of monocytes to the endothelium, leading to vascular dysfunction [43]. Hyperglycemia is the most potent driver of Set7 upregulation, and the expression of Set7 in circulating monocytes may represent a potential marker of vascular damage. Targeting this chromatin-modifying enzyme may represent a novel therapeutic approach [43].
High glucose also induces DNA demethylation of the superoxide dismutase 2 (SOD2) promoter through TET2 upregulation, leading to reduced SOD2 expression, increased oxidative stress and vascular dysfunction [39].
Hyperacetylation of histone H3 at lysine residues 9 and 14 (H3K9/K14) coexisting with DNA hypomethylation of CpG islands was observed in response to hyperglycemia in human vascular cells on multiple gene promoters, including heme oxygenase 1 (HMOX1), IL-8, matrix metalloproteinase 1 (MMP1) and 10 (MMP10) and cysteine/glutamate transporter (SCL7A11) [44]. Chromatin immunoprecipitation assays in monocytes demonstrated that diabetic patients present an increased acetylation of histone H3 at lysine residues 9 and 14 and of histone H4 at lysine residues 5, 8 and 12 of cyclooxygenase 2 (COX2), TNFα and MCP-1 gene promoters, causing enhanced inflammatory genes transcription and monocyte activation [45]. These genomic regions with high amounts of H3K9/14 histone tail hyperacetylation represent 0.3% of the genome in cells cultured in hyperglycemic conditions [44].
Hyperglycemia also induces TET-2 mediated DNA demethylation changes that are involved in the differentiation of the vessel smooth muscle cells in a phenotype characterized by loss of contractility and increased proliferation and secretion of extracellular matrix proteins that promote atherosclerosis. TET2 knockdown determines the reduced expression of vascular smooth muscle cells (VSMCs) contractile genes (such as MYOCD, SRF and MYH11 genes), increased the expression of synthetic phenotype markers, such as KLF4, KLF5 and OPN genes and the increased proliferation of human coronary artery VSMCs [46]. A U.K. Prospective Diabetes Study showed that end-organ effects continued to operate >5 years after the patients had returned to their usual level of glycemic control because of the “hyperglycemic memory” or “legacy effect” [47].
Transient hyperglycemia and the relative induced epigenetic modifications are able to determine a persistent activation of genes harmful to the cardiovascular system, even when glycemic control is reached and maintained. Human aortic endothelial cells exposed for 7 days to high glucose presented lower DNA methylation levels for the SIRT6 promoter with increased expression of SIRT6 and TET2; these epigenetic changes persisted after 48 h of glucose normalization [48]. The increased SIRT6 expression following high-glucose exposure might be a compensatory response to hyperglycemia-induced damage, since SIRT6 is involved in the DNA-damage repair system and protects against endothelial dysfunction and vascular inflammation [49].
Epigenetic changes in the promoter of NF-κB subunit p65 (increased H3K4m1 by the histone methyltransferase Set7) in aortic endothelial cells induced by transient hyperglycemia (16 h) persist for at least 6 days of subsequent normal glycemia, as do NF-κB-induced increases in MCP-1 and VCAM-1 expression [50]. H3K4 methylation of NF-κB p65 subunit also provides a mechanism for epigenetic memory in macrophages [51].
Short-term (72 h) high glucose stimulation induces persistent downregulation of deacetylase SIRT1 as well as the upregulation of acetyltransferase p300, leading to sustained hyperacetylation (at K382) and activation of p53 and subsequent p53/p21-mediated senescent “memory” [52].
Hyperglycemia is not the only trigger of epigenetic modifications. AGEs directly reduce SIRT1 expression, leading to an increased acetylation process and to the activation of p53, which is responsible for apoptosis and endothelial dysfunction [53]. Furthermore, the products of oxidative processes induced by hyperglycemia determine p66Shc promoter DNA demethylation and GCN5-mediated histone 3 hyperacetylation. These epigenetic modifications lead to p66Shc overexpression, which supports ROS production and antioxidant manganese superoxide dismutase downregulation, resulting in unchallenged ROS accumulation in the vascular endothelium and endothelial dysfunction [54].
ROS production activates PKCβII, which in turn maintains elevated p66Shc levels, promoting sustained ROS-dependent epigenetic changes. Hyperglycemia-mediated ROS overproduction is indeed considered the major driver of glycemic memory in the vasculature [35]. Some authors hypothesized a two-stage mechanism for hyperglycemic memory: an initial induction phase due to hyperglycemia-induced increased ROS production and a perpetuation phase, since ROS induces mutations in the mitochondrial DNA with the encoding of defective electron transport chain subunits that cause increased ROS production by the electron transport chain also at physiologic concentrations of glucose and glucose-derived reducing equivalents [20].
SIRT1 deacetylates H3 histones at the human endothelial p66Shc promoter to suppress the transcription of ROS and promote transcription of SOD; therefore, SIRT1 prevents hyperglycemia-induced endothelial dysfunction and avoids hyperglycemic memory [55]. Adipose tissue from insulin-resistant subjects presents DNA hypermethylation of different genes, such as FTO (associated with increased risk of obesity and cardiovascular diseases) [56] and IGF2 (associated with a higher triglyceride/HDL ratio and increased metabolic risk in children) genes [57].
Low-grade chronic inflammation and insulin resistance induce histone deacetylase 3 (HDAC3) activity and expression in peripheral blood mononuclear cells [58]. A HDAC3 increase is associated with a decreased transcriptional level of DCB1, which negatively regulates HDAC3. HDAC3 activity correlates with increased TNFα, IL-6, MCP-1 and reduced SIRT1, which causes macrophage recruitment and infiltration into adipose tissue [58]. In obesity-induced insulin resistance/T2DM, the elevated levels of saturated fatty acids (SFAs) enhance DNA methyltransferase 3b activity, leading to the methylation of the PPRγ promoter and triggering the macrophage phenotype switch from anti-inflammatory (M2) to pro-inflammatory (M1) [59].
References
- Poznyak, A.; Grechko, A.V.; Poggio, P.; Myasoedova, V.A.; Alfieri, V.; Orekhov, A.N. The Diabetes Mellitus Atherosclerosis Connection: The Role of Lipid and Glucose Metabolism and Chronic Inflammation. Int. J. Mol. Sci. 2020, 21, 1835.
- Bierman, E.L. George Lyman Duff Memorial Lecture. Atherogenesis in diabetes. Arter. Thromb. A J. Vasc. Biol. 1992, 12, 647–656.
- Pyorala, K.; Laakso, M.; Uusitupa, M. Diabetes and atherosclerosis: An epidemiologic view. Diab. Metab. Rev. 1987, 3, 463–524.
- Virchow, R. Cellular Pathology; John Churchill: London, UK, 1858.
- Lee, H.M.; Kim, J.J.; Kim, H.J.; Shong, M.; Ku, B.J.; Jo, E.K. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes 2013, 62, 194–204.
- Hess, K.; Grant, P.J. Inflammation and thrombosis in diabetes. Thromb. Haemost. 2011, 105, S43–S54.
- Jarrett, R.J. Type 2 (non-insulin-dependent) diabetes mellitus and coronary heart disease: Chicken, egg, or neither? Diabetologia 1984, 26, 99–102.
- Stern, M.P. Diabetes and Cardiovascular Disease: The “Common Soil” Hypothesis. Diabetes 1995, 44, 369–374.
- De Rosa, S.; Arcidiacono, B.; Chiefari, E.; Brunetti, A.; Indolfi, C.; Foti, D.P. Type 2 Diabetes Mellitus and Cardiovascular Disease: Genetic and Epigenetic Links. Front. Endocrinol. 2018, 9, 2.
- Pfohl, M.; Koch, M.; Enderle, M.D.; Kühn, R.; Füllhase, J.; Karsch, K.R.; Häring, H.U. Paraoxonase 192 Gln/Arg gene polymorphism, coronary artery disease, and myocardial infarction in type 2 diabetes. Diabetes 1999, 48, 623–627.
- Garin, M.C.; James, R.W.; Dussoix, P.; Blanché, H.; Passa, P.; Froguel, P.; Ruiz, J. Paraoxonase polymorphism Met-Leu54 is associated with modified serum concentrations of the enzyme. A possible link between the paraoxonase gene and increased risk of cardiovascular disease in diabetes. J. Clin. Investig. 1997, 99, 62–66.
- Jones, D.; Prior, S.; Tang, T.; Bain, S.; Hurel, S.; E Humphries, S.; Stephens, J. Association between the rs4880 superoxide dismutase 2 (C>T) gene variant and coronary heart disease in diabetes mellitus. Diabetes Res. Clin. Pract. 2010, 90, 196–201.
- Bacci, S.; Menzaghi, C.; Ercolino, T.; Ma, X.; Rauseo, A.; Salvemini, L.; Vigna, C.; Fanelli, R.; Di Mario, U.; Doria, A.; et al. The +276 G/T Single Nucleotide Polymorphism of the Adiponectin Gene Is Associated with Coronary Artery Disease in Type 2 Diabetic Patients. Diabetes Care 2004, 27, 2015–2020.
- Soccio, T.; Zhang, Y.-Y.; Bacci, S.; Mlynarski, W.; Placha, G.; Raggio, G.; Di Paola, R.; Marucci, A.; Johnstone, M.T.; Gervino, E.V.; et al. Common Haplotypes at the Adiponectin Receptor 1 (ADIPOR1) Locus Are Associated with Increased Risk of Coronary Artery Disease in Type 2 Diabetes. Diabetes 2006, 55, 2763–2770.
- El-Lebedy, D.; Raslan, H.M.; Mohammed, A.M. Apolipoprotein E gene polymorphism and risk of type 2 diabetes and cardiovascular disease. Cardiovasc. Diabetol. 2016, 15, 12.
- Qi, L.; Parast, L.; Cai, T.; Powers, C.; Gervino, E.V.; Hauser, T.H.; Hu, F.B.; Doria, A. Genetic Susceptibility to Coronary Heart Disease in Type 2 Diabetes: 3 Independent Studies. J. Am. Coll. Cardiol. 2011, 58, 2675–2682.
- De Rosa, S.; Chiefari, E.; Salerno, N.; Ventura, V.; D’Ascoli, G.L.; Arcidiacono, B.; Ambrosio, G.; Bilotta, F.L.; Torella, D.; Foti, D.; et al. HMGA1 is a novel candidate gene for myocardial infarction susceptibility. Int. J. Cardiol. 2017, 227, 331–334.
- Ferrannini, G.; Manca, M.L.; Magnoni, M.; Andreotti, F.; Andreini, D.; Latini, R.; Maseri, A.; Maggioni, A.P.; Ostroff, R.M.; Williams, S.A.; et al. Coronary Artery Disease and Type 2 Diabetes: A Proteomic Study. Diabetes Care 2020, 43, 843–851.
- Kolluru, G.K.; Bir, S.C.; Kevil, C.G.; Calvert, J.W. Endothelial Dysfunction and Diabetes: Effects on Angiogenesis, Vascular Remodeling, and Wound Healing. Int. J. Vasc. Med. 2012, 2012, 918267.
- Nishikawa, T.; Edelstein, D.; Brownlee, M. The missing link: A single unifying mechanism for diabetic complications. Kidney Int. 2000, 58 (Suppl. 77), S26–S30.
- Weykamp, C. HbA1c: A Review of Analytical and Clinical Aspects. Ann. Lab. Med. 2013, 33, 393–400.
- Lachin, J.M.; Orchard, T.J.; Nathan, D.M.; for the DCCT/EDIC Research Group. Update on Cardiovascular Outcomes at 30 Years of the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Study. Diabetes Care 2013, 37, 39–43.
- Kuusisto, J.; Mykkanen, L.; Pyorala, K.; Laakso, M. NIDDM and its metabolic control predict heart disease in elderly subjects. Diabetes 1994, 43, 960–967.
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced glycation end products: Sparking the development of diabetic vascular injury. Circulation 2006, 114, 597–605.
- Prandi, F.R.; Evangelista, I.; Sergi, D.; Palazzuoli, A.; Romeo, F. Mechanisms of cardiac dysfunction in diabetic cardiomyopathy: Molecular abnormalities and phenotypical variants. Heart Fail. Rev. 2022. Epub ahead of print.
- Rajbhandari, J.; Fernandez, C.J.; Agarwal, M.; Yeap, B.X.Y.; Pappachan, J.M. Diabetic heart disease: A clinical update. World J. Diabetes 2021, 12, 383–406.
- Aronson, D. Cross-linking of glycated collagen in the pathogenesis of arterial and myocardial stifening of aging and diabetes. J. Hypertens 2003, 21, 3–12.
- Maguire, E.M.; Pearce, S.W.A.; Xiao, Q. Foam cell formation: A new target for fighting atherosclerosis and cardiovascular disease. Vascul. Pharmacol. 2019, 112, 54–71.
- Zeadin, M.G.; Petlura, C.I.; Werstuck, G.H. Molecular mechanisms linking diabetes to the accelerated development of atherosclerosis. Can. J. Diabetes 2013, 37, 345–350.
- Brealey, D.; Singer, M. Hyperglycemia in Critical Illness: A Review. J. Diabetes Sci. Technol. 2009, 3, 1250–1260.
- Reaven, G.M.; Lithell, H.; Landsberg, L. Mechanisms of disease: Hypertension and associated metabolic abnormalities. The role of insulin resistance and the sympathoadrenal system. N. Engl. J. Med. 1996, 334, 374–381.
- Carey, D.G.; Jenkins, A.B.; Campbell, L.V.; Freund, J.; Chisholm, D.J. Abdominal fat and insulin resistance in normal and overweight women: Direct measurements reveal a strong relationship in subjects at both low and high risk of NIDDM. Diabetes 1996, 45, 633–638.
- Wu, C.; Morris, J.R. Genes, genetics, and epigenetics: A correspondence. Science 2001, 293, 1103–1105.
- Alaskhar Alhamwe, B.; Khalaila, R.; Wolf, J.; Von Bülow, V.; Harb, H.; Alhamdan, F.; Hii, C.S.; Prescott, S.L.; Ferrante, A.; Renz, H.; et al. Histone modifications and their role in epigenetics of atopy and allergic diseases. Allergy Asthma Clin. Immunol. 2018, 14, 39.
- Keating, S.; Plutzky, J.; El-Osta, A. Epigenetic Changes in Diabetes and Cardiovascular Risk. Circ. Res. 2016, 118, 1706–1722.
- Gao, C.; Huang, W.; Kanasaki, K.; Xu, Y. The Role of Ubiquitination and Sumoylation in Diabetic Nephropathy. BioMed Res. Int. 2014, 2014, 160692.
- Zhu, D.; Zhang, Y.; Wang, S. Histone citrullination: A new target for tumors. Mol. Cancer 2021, 20, 90.
- Manolopoulos, V.G.; Ragia, G.; Tavridou, A. Pharmacogenomics of oral antidiabetic medications: Current data and pharmacoepigenomic perspective. Pharmacogenomics 2011, 12, 1161–1191.
- Scisciola, L.; Rizzo, M.R.; Cataldo, V.; Fontanella, R.A.; Balestrieri, M.L.; D’Onofrio, N.; Marfella, R.; Paolisso, G.; Barbieri, M. Incretin drugs effect on epigenetic machinery: New potential therapeutic implications in preventing vascular diabetic complications. FASEB J. 2020, 34, 16489–16503.
- Brasacchio, D.; Okabe, J.; Tikellis, C.; Balcerczyk, A.; George, P.; Baker, E.K.; Calkin, A.C.; Brownlee, M.; Cooper, M.E.; El-Osta, A. Hyperglycemia Induces a Dynamic Cooperativity of Histone Methylase and Demethylase Enzymes Associated With Gene-Activating Epigenetic Marks That Coexist on the Lysine Tail. Diabetes 2009, 58, 1229–1236.
- Huang, W.; Xu, L.; Zhou, X.; Gao, C.; Yang, M.; Chen, G.; Zhu, J.; Jiang, L.; Gan, H.; Gou, F.; et al. High glucose induces activation of NF-κB inflammatory signaling through IκBα sumoylation in rat mesangial cells. Biochem. Biophys. Res. Commun. 2013, 438, 568–574.
- Marfella, R.; Di Filippo, C.; Portoghese, M.; Siniscalchi, M.; Martis, S.; Ferraraccio, F.; Guastafierro, S.; Nicoletti, G.; Barbieri, M.; Coppola, A.; et al. The ubiquitin–proteasome system contributes to the inflammatory injury in ischemic diabetic myocardium: The role of glycemic control. Cardiovasc. Pathol. 2009, 18, 332–345.
- Paneni, F.; Costantino, S.; Battista, R.; Castello, L.; Capretti, G.; Chiandotto, S.; Scavone, G.; Villano, A.; Pitocco, D.; Lanza, G.; et al. Adverse epigenetic signatures by histone methyltransferase Set7 contribute to vascular dysfunction in patients with type 2 diabetes mellitus. Circ. Cardiovasc. Genet. 2015, 8, 150–158.
- Pirola, L.; Balcerczyk, A.; Tothill, R.W.; Haviv, I.; Kaspi, A.; Lunke, S.; Ziemann, M.; Karagiannis, T.; Tonna, S.; Kowalczyk, A.; et al. Genome-wide analysis distinguishes hyperglycemia regulated epigenetic signatures of primary vascular cells. Genome Res. 2011, 21, 1601–1615.
- Miao, F.; Gonzalo, I.G.; Lanting, L.; Natarajan, R. In Vivo Chromatin Remodeling Events Leading to Inflammatory Gene Transcription under Diabetic Conditions. J. Biol. Chem. 2004, 279, 18091–18097.
- Liu, R.; Jin, Y.; Tang, W.H.; Qin, L.; Zhang, X.; Tellides, G.; Hwa, J.; Yu, J.; Martin, K.A. Ten-Eleven Translocation-2 (TET2) Is a Master Regulator of Smooth Muscle Cell Plasticity. Circulation 2013, 128, 2047–2057.
- Chalmers, J.; Cooper, M.E. UKPDS and the legacy effect. N. Engl. J. Med. 2008, 359, 1618–1620.
- Scisciola, L.; Rizzo, M.R.; Marfella, R.; Cataldo, V.; Fontanella, R.A.; Boccalone, E.; Paolisso, G.; Barbieri, M. New insight in molecular mechanisms regulating SIRT6 expression in diabetes: Hyperglycaemia effects on SIRT6 DNA methylation. J. Cell. Physiol. 2020, 236, 4604–4613.
- McCord, R.A.; Michishita, E.; Hong, T.; Berber, E.; Boxer, L.; Kusumoto, R.; Guan, S.; Shi, X.; Gozani, O.; Burlingame, A.L.; et al. SIRT6 stabilizes DNA-dependent Protein Kinase at chromatin for DNA double-strand break repair. Aging 2009, 1, 109–121.
- El-Osta, A.; Brasacchio, D.; Yao, D.; Pocai, A.; Jones, P.L.; Roeder, R.G.; Cooper, M.E.; Brownlee, M. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J. Exp. Med. 2008, 205, 2409–2417.
- Ostuni, R.; Piccolo, V.; Barozzi, I.; Polletti, S.; Termanini, A.; Bonifacio, S.; Curina, A.; Prosperini, E.; Ghisletti, S.; Natoli, G. Latent Enhancers Activated by Stimulation in Differentiated Cells. Cell 2013, 152, 157–171.
- Zhang, E.; Guo, Q.; Gao, H.; Xu, R.; Teng, S.; Wu, Y. Metformin and Resveratrol Inhibited High Glucose-Induced Metabolic Memory of Endothelial Senescence through SIRT1/p300/p53/p21 Pathway. PLoS ONE 2015, 10, e0143814.
- Li, P.; Zhang, L.; Zhou, C.; Lin, N.; Liu, A. Sirt 1 activator inhibits the AGE-induced apoptosis and p53 acetylation in human vascular endothelial cells. J. Toxicol. Sci. 2015, 40, 615–624.
- Paneni, F.; Volpe, M.; Lüscher, T.F.; Cosentino, F. SIRT1, p66Shc, and Set7/9 in Vascular Hyperglycemic Memory. Diabetes 2013, 62, 1800–1807.
- Zhou, S.; Chen, H.-Z.; Wan, Y.-Z.; Zhang, Q.-J.; Wei, Y.-S.; Huang, S.; Liu, J.-J.; Lu, Y.-B.; Zhang, Z.-Q.; Yang, R.-F.; et al. Repression of P66Shc Expression by SIRT1 Contributes to the Prevention of Hyperglycemia-Induced Endothelial Dysfunction. Circ. Res. 2011, 109, 639–648.
- Bell, C.G.; Finer, S.; Lindgren, C.M.; Wilson, G.A.; Rakyan, V.K.; Teschendorff, A.E.; Akan, P.; Stupka, E.; Down, T.A.; Prokopenko, I.; et al. Integrated Genetic and Epigenetic Analysis Identifies Haplotype-Specific Methylation in the FTO Type 2 Diabetes and Obesity Susceptibility Locus. PLoS ONE 2010, 5, e14040.
- Deodati, A.; Inzaghi, E.; Liguori, A.; Puglianiello, A.; Germani, D.; Brufani, C.; Fintini, D.; Cappa, M.; Barbetti, F.; Cianfarani, S. IGF2Methylation Is Associated with Lipid Profile in Obese Children. Horm. Res. Paediatr. 2013, 79, 361–367.
- Sathishkumar, C.; Prabu, P.; Balakumar, M.; Lenin, R.; Prabhu, D.; Anjana, R.M.; Mohan, V.; Balasubramanyam, M. Augmentation of histone deacetylase 3 (HDAC3) epigenetic signature at the interface of proinflammation and insulin resistance in patients with type 2 diabetes. Clin. Epigenet. 2016, 8, 125.
- Yang, X.; Wang, X.; Liu, D.; Yu, L.; Xue, B.; Shi, H. Epigenetic Regulation of Macrophage Polarization by DNA Methyltransferase 3b. Mol. Endocrinol. 2014, 28, 565–574.
More
Information
Subjects:
Cardiac & Cardiovascular Systems
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
3.0K
Revisions:
3 times
(View History)
Update Date:
13 May 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No