Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Davide Bassani | -- | 5990 | 2022-05-10 23:34:36 | | | |
| 2 | Davide Bassani | Meta information modification | 5990 | 2022-05-10 23:40:17 | | | | |
| 3 | Jason Zhu | -170 word(s) | 5820 | 2022-05-11 03:30:57 | | | | |
| 4 | Davide Bassani | + 1351 word(s) | 7171 | 2022-05-11 21:09:53 | | | | |
| 5 | Jason Zhu | -86 word(s) | 7085 | 2022-05-12 04:08:07 | | | | |
| 6 | Jason Zhu | -3 word(s) | 7082 | 2022-05-12 04:11:05 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Bassani, D.; Moro, S.; Pavan, M.; Federico, S.; Spalluto, G.; Sturlese, M. G Protein-Coupled Receptors in Amyotrophic Lateral Sclerosis. Encyclopedia. Available online: https://encyclopedia.pub/entry/22788 (accessed on 09 August 2026).
Bassani D, Moro S, Pavan M, Federico S, Spalluto G, Sturlese M. G Protein-Coupled Receptors in Amyotrophic Lateral Sclerosis. Encyclopedia. Available at: https://encyclopedia.pub/entry/22788. Accessed August 09, 2026.
Bassani, Davide, Stefano Moro, Matteo Pavan, Stephanie Federico, Giampiero Spalluto, Mattia Sturlese. "G Protein-Coupled Receptors in Amyotrophic Lateral Sclerosis" Encyclopedia, https://encyclopedia.pub/entry/22788 (accessed August 09, 2026).
Bassani, D., Moro, S., Pavan, M., Federico, S., Spalluto, G., & Sturlese, M. (2022, May 10). G Protein-Coupled Receptors in Amyotrophic Lateral Sclerosis. In Encyclopedia. https://encyclopedia.pub/entry/22788
Bassani, Davide, et al. "G Protein-Coupled Receptors in Amyotrophic Lateral Sclerosis." Encyclopedia. Web. 10 May, 2022.
Copy Citation
Amyotrophic lateral sclerosis (ALS) is a degenerating disease involving the motor neurons, which causes a progressive loss of movement ability, usually leading to death within 2 to 5 years from the diagnosis. Much effort has been put into research for an effective therapy for its eradication, but still, no cure is available. The only two drugs approved for this pathology, Riluzole and Edaravone, are onlyable to slow down the inevitable disease progression. As assessed in the literature, drug targets such as protein kinases have already been extensively examined as potential drug targets for ALS, with some molecules already in clinical trials.
amyotrophic
sclerosis
ALS
GPCR
review
drug
therapy
1. Introduction
Amyotrophic Lateral Sclerosis (ALS, also referred to as “motor neuron disease”) indicates a clinical situation in which the motor neurons of patients undergo a progressive loss in their function and number [1]. This type of neuronal cell, whose cell body is localized in the motor cortex, the brainstem, and the spinal cord, is responsible for the innervation and the control of muscle fibers, essential for voluntary muscle contraction [2]. Their loss has very important consequences on the patient’s life, firstly impairing the ability to chew and walk, then to speak and to move, until even the ability to breath is affected, leading, after 2–5 years, to death due to respiratory failure [3]. ALS can be classified into two main types, “sporadic ALS” (the great majority of all cases), which has no known cause and typically has its onset between the ages of 58 and 63 years, and “familial ALS” (about 5–10% of cases), which is linked to genetic factors, and has its onset between the ages of 47 and 52 years [4].
In both scenarios, the pathology starts with the manifestation of muscle weakness and atrophy, with methods and timing very variable based on the patient and on the parts of the motor neurons that are affected first [5]. Indeed, a classification of the onset of the pathology can be made with reference to the site of its onset. For two-thirds of patients, the limb muscles are affected first (“spinal ALS”), with manifestations mainly in the distal muscles of the dominant hand for the upper limb and in the hamstrings for the lower limb. For the greater part of the remaining patients, the bulbar muscles represent the onset site (“bulbar ALS”), and in this case, dysphagia and chewing problems represent the first manifestations of the pathology [6].
The next steps of the disease involve the progressive spreading of the neurodegeneration process to the unaffected motor neurons, causing an increasing worsening in the patient’s daily life, making activities such as eating and walking continuously more difficult and leading to their complete loss. The final and worst clinical scenario has its onset when the respiratory function is significantly affected, progressively increasing the risk of respiratory failure, which is the main cause of death due to ALS [7].
Even if much effort has been made among both academic and industrial scientific groups, no cure has yetemerged for ALS. Riluzole [8] and the recently FDA-approved drug Edaravone [9] (both represented in Figure 1) constitute the only two small molecules used for the ALS treatment, and only succeed in slowing down the disease’s progression [10].
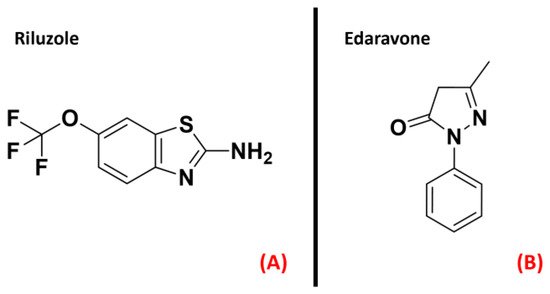
Figure 1. The chemical structures of Riluzole (A) and Edaravone (B), which are the only two small-molecule drugs approved currently for ALS treatment.
Such a neurodegenerating process, affecting 1.75–3 people per 100,000 [11], has, in the great majority of cases, no known cause [12], making it even harder to design a therapy for this disease. From a biochemical point of view, the hallmark of ALS is considered to be the presence of inclusion bodies in the cytoplasm of motor neurons. These aggregates are formed by the TAR DNA-binding protein 43 (TDP-43) [13], a protein involved in several important physiological functions such as DNA repair, splicing, and transcriptional regulation. Even if its main localization site is the nucleus, processes such as its hyperphosphorylation or the mutation of its gene (TARDBP) lead to its aggregation in the cytoplasm [14]. This mislocalization directly causes the dysregulation of several cellular events related to RNA metabolism, DNA replication, and oxidative stress management, leading to the loss of the motor neurons affected [15]. Other molecular targets that have been demonstrated to be important for ALS onset and progression are superoxide dismutase (SOD1) [16] and DNA/RNA-binding protein FUS/TLS (FUused in Sarcoma/Translocated in LipoSarcoma, also called “FUS”) [17], which appear to be mutated in the patients.
The knowledge that hyperphosphorylation of TDP-43 is one of the main processes leading to its aggregation has led the scientific community to devote some effort to identifying the protein kinases responsible for such processes, in order to find proper inhibitors for such species [18]. Recent work by Guo et al. goes deep in the examination of the involvement of kinases in ALS progression, enucleating species such as CK1, ERK, GSK3β, and JAK3 as promising targets for the treatment of this neurodegenerating disease [19]. Another article by Palomo et al. gives an exhaustivepanoramic view of the protein kinase inhibitors currently in clinical trials for ALS treatment [20]. Riluzole has proven to increase life expectancy by about 2–3 months [21], and even if its main target still remains the NMDA receptor, recent work by Bissaro et al. suggested that this mechanism could be due to its action on the delta isoform of CK1 [22].
Many molecular candidates (both new chemical entities and compounds coming from repurposing strategies) are nowadays in clinical trials for ALS [23], acting on different biological pathways, with the common aim being to restore the neuronal health status in the affected patients, possibly trying to go in the direction to find a proper cure for this pathology [24].
Even if some effort has been directed toward trying to highlight the role of protein kinases in ALS progression, this has not been recently or extensively done with respect to G-protein coupled receptors, biological actors which have been demonstrated to be detrimental to neuronal and physiological conditions.It is important to remember that ALS is a non-cell-autonomous disease, which means that the neuronal damage characterizing the pathology is caused by aberrant processes also happening outside the neurons themselves. Indeed, ALS progression has been demonstrated to be strongly related to glial cell dysregulation (mainly microglia and astrocytes) [25]. GPCRs are very widely expressed proteins in the human organism [26], and so a beneficial effect could also be obtained by targeting extraneuronal receptors, which could trigger biological processes that, in the overall scenario, could mitigate if not reverse the disease progression.
GPCRs are membrane receptors and constitute one of the main protein families encoded by human genes, with more than 800 members already identified [27], divided intosixdifferent classes (identified alphabetically with letters from “A” to “F”) based on their similarities in sequence and function. They all share a common architecture formed of a seven-α-helixtransmembrane domain (usually referred to as “7-TM”), an extracellular N-terminal domain, and an intracellular C-terminal domain. These proteins exert their roles by coupling with an intracellular messenger called “heterotrimeric G protein”, which is formed by α, β, and γ subunits, and interacts with different intracellular partners based on its type. The α subunit is displaced from the βγ-complex upon GPCR–ligand binding, and its fate depends on its Gαfamily belonging. Indeed, activated Gi/0 proteins inhibit adenylyl cyclase (AC), reducing the production of the second messenger cyclic adenosine monophosphate (cAMP); Gs, conversely, activates adenylyl cyclase, and Gqαsubunits activate phospholipase C (PLC), leading to an increase in Ca2+ influx in the cytoplasm [28].The physiological roles of GPCRsinclude homeostasis modulation, mood balancing, immune system regulation, neuronal plasticity, and many more [29]. The goal of the present work is to give a panoramic view of the GPCRs which have been linked to ALS onset and progression, presenting what has already been done to modulate their action, and highlighting new potential therapeutical scenarios.
2. GPCRs Involved in ALS
2.1. Puringergic Receptors P2Y and Adenosine Receptor A2AAR
Purinergic receptors are a peculiar class of membrane receptors, sensitive to a wide series of purinergic ligands such as ATP, ADP, UTP, UDP, UDP-glucose, and adenosine. The kind of molecules interacting with them defines their classification intoone of the three subfamilies forming this class. The first group, called “P1 receptors”, is formed of GPCRs activated upon adenosine binding (and for these reasons are also known as “adenosine receptors”), while “P2Y receptors” are GPCRs that can bind to ATP, UDP, and their diphosphate analogs ADP and UDP (with the addition of UDP-glucose). The last subfamily, named “P2X receptors”, are ligand-gated ion channels exclusively sensitive to ATP [30]. Being the P2X family not formed by GPCRs, evaluations will focus on the first two families of receptors. The “P1” subfamily, more commonly referred to as adenosine receptors (ARs), is a group of purinergic class AGPCRs divided into four subtypes, A1AR, A2AAR, A2BAR, and A3AR, each involved in many different physiological processes. While the functions of A2BAR and A3AR are mainly related to the circulatory, immune and respiratory systems, the A1AR and A2AAR proteins are importantly present in the central nervous system (CNS) [31]. Moreover, the A1AR and the A2AAR receptors have been demonstrated to play a crucial role in neuroprotection, neuronal survival, and neuroinflammation [32]. A study from Vincenzi et al. reported an upregulation of A2AAR receptors in the lymphocytes of people affected by ALS [33], while Yoshida et al. measured adenosine levels in the cerebrospinal fluid of ALS patients, finding out that these were significantly higher with respect to the control subjects [34]. Unexpectedly, treatment with the A2AAR antagonist caffeine (Figure 2, panel A), which is usually referred to as a protective agent against Alzheimer’s Disease (AD) and Parkinson’s Disease (PD), was demonstrated to shorten the survival of ALS-affected SOD1G93A mice, a well-known experimental model for ALS (Potenza et al.) [35], even if some explanation for this phenomenon can be provided by the non-selectivity of caffeine [36]. Indeed, Ng et al. showed that suppression of A2AAR signaling delays the progression of ALS in the same SOD1G93A mouse model [37]. This is in accordance with the study of Mojsilovic-Petrovic et al., which demonstrated that A2AAR inhibitors (such as the non-selective enprofylline (Figure 2, panel B), and the A2AAR-selective KW-6002 (Figure 2, panel C, also called Istradefylline) protect motor neurons from toxic insult, highlighting the beneficial effects of such activity for ALS patients [38].
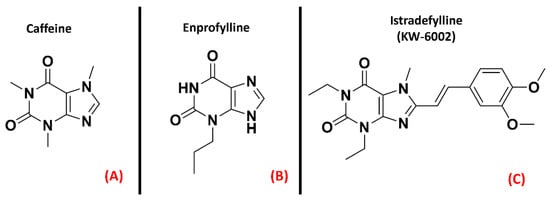
Figure 2. The chemical structures of the non-selective adenosine receptors antagonists caffeine (A) and Enprofylline (B), and the selective A2AAR antagonist Istradefylline (C).
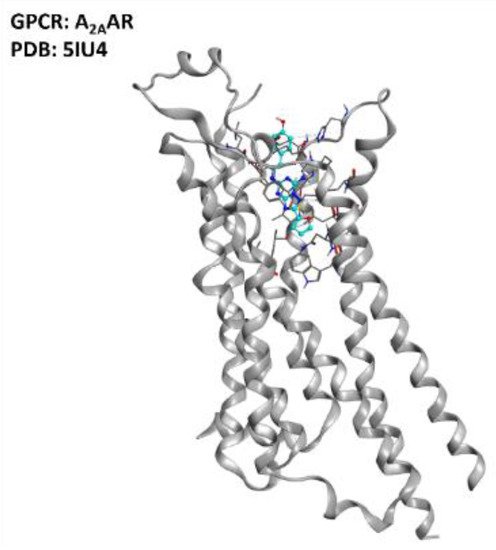
Despite all this evidence, a study from Liu et al. showed that A2AAR activation, suppressing AMPK activation, suppressed TDP-43 mislocalization [39]. The multifactorial nature of ALS makes it very difficult to define sharply whether agonism or antagonism of the A2AAR receptor has the best risk/benefit ratio, but the literature clearly defines this GPCR (represented in Figure 3) as one of the promising targets for ALS treatment.
The second subfamily of purinergic receptors, “P2Y”,is present in a great variety of human tissues, but their main biological roles are identifiable in blood clotting, vasodilatation, and immune response [43]. The P2Y family comprises eight different isoforms, among which P2Y12 has gained the interest of the scientific community for its role in neuroinflammation, as addressed by Morillas et al. [44] and Amadio et al. [45]. Jacobson et al. recently highlighted the proinflammatory effect of P2Y agonists, reporting that antagonizing this class of GPCRs could be considered a way of treating inflammatory conditions [46]. Even if inhibitors of the P2Y12 isoform are already marketed as antiplatelet drugs (e.g., Clopidogrel, Prasugrel, Ticagrelor, all represented in Figure 4), Jacobson et al. pointed out that there is a lack of selective and versatile P2Y ligands for each subtype, meaning that the drug discovery process is still very active in this specific field.
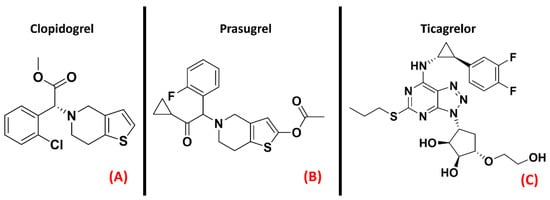
Figure 4. The chemical structures of the selective and irreversible P2Y12 receptor antagonists Clopidogrel (A) and Prasugrel (B). Ticagrelor, a selective, reversible, allosteric P2Y12 receptor antagonist, is also reported (C).
Specifically, D’Ambrosi et al. reported an upregulation of P2Y6 receptors in the microglia SOD1 mutant models of ALS, also remembering that this phenomenon is associated with brain damage [47]. Moreover, P2Y12 (represented in Figure 5) is upregulated in spinal cord microglia upon nerve injury, as pointed out by Kobayashi et al. [48]. Converging information is provided by a study from Moore et al. [49], further confirming P2Y12 as a potential target for modulating neuroinflammation and neuronal damage. The data currently available help in suggesting the practical possibility of ALS regulation through purinergic receptor modulation, and this will be realizable as soon as proper inhibitors can be designed, potentially avoiding the non-desired antiplatelet effect of these molecules.
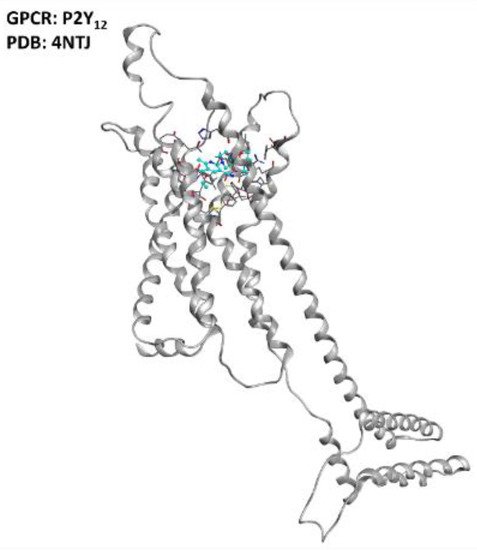
Figure 5. Representation of the structure of the P2Y12 receptor (sourced from the Protein Data Bank, PDB code: 4NTJ [43], method: X-ray diffraction, resolution: 2.62 Å). The image was created and rendered with MOE.
2.2. Chemokine Receptors
Chemokine receptors constitute a group of about 20 classes of GPCRs found mainly on the surface of leukocytes, which respond to specific ligands to control chemotaxis. The ligands for these proteins are called chemokines and are a peculiar kind of cytokine used for inducing a directional movement of certain types of cells, such as epithelial and immune ones. The chemokines, as well as the receptors they act on, can be divided into four families, namely CC (e.g., chemokine CCL4), CXC (e.g., chemokine CXCL8, also known as IL-8), XC (e.g., XCL1), and CX3C (of which the only member today is CX3CL1, also called neurotactin) [50]. The activation of chemokine receptors leads to Ca2+ influx and cell mobilization [51]. Several of these receptors are important in the progression of motor neuron damage. La Cognata et al. highlighted an upregulation of CXCR2 in both sporadic ALS patients and SOD1G93A mice, showing that by treating the mouse models with the CXCR2 allosteric inhibitor Reparixin (Figure 6, panel B), the neuromuscular function of the subjects was improved [52]. Another interesting paper published by Rabinovich-Nikitin et al. highlighted the benefits in terms of lifespan and motor function obtained on SOD1G93A mouse models through the administration of the CXCR4 antagonist AMD3100 (also known as “Plerixafor”, Figure 6, panel A) [53].
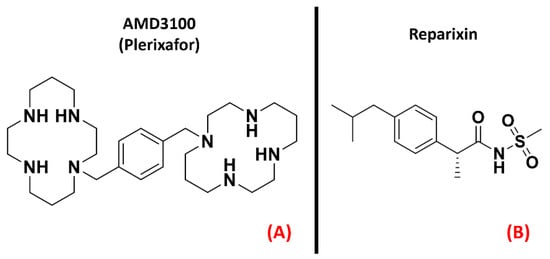
Figure 6. The chemical structures of the CXCR4 antagonist AMD3100 (also known as “Plerixafor”, (A) and the inhibitor of CXCR1 and CXCR2 known as Reparixin (B).
Several scientific works have reported an increase in circulating chemokines and cytokines in ALS patients, as recently detailed by Liu et al. [54], and the upregulation of chemokine receptors CXCR3, CXCR4, and CCR2 was also highlighted in the pathology of interest by Perner et al. [55], who also proposed CXCR3 and its ligands as possible therapeutic targets for ALS. These scientific works converge in addressing chemokine receptor modulation as a possibility for ALS treatment, focusing on the antagonism of certain isoforms. The three-dimensional structures of CXCR2, CXCR3, and CXCR4 are represented in Figure 7.
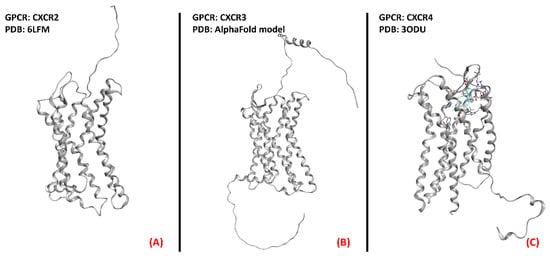
Figure 7. Representation of the three-dimensional structures of the chemokine receptors that could be considered for ALS treatment. CXCR2 (A) receptor (sourced from the Protein Data Bank, PDB code: 6LFM [56], method: cryo-EM, resolution: 2.50 Å), CXCR3 (B) (with no experimentally resolved structure available, the model from the AlphaFold [57] database is presented), and CXCR4 (C) receptor (sourced from the Protein Data Bank, PDB code: 3ODU [58], method: X-ray diffraction, resolution: 2.50 Å).The images were created and rendered with MOE.
2.3. Angiotensin II Receptors (ATRs)
Angiotensin II receptors (ATRs) are a group of GPCRs that have gained fame for their importance in the therapy of hypertension. Indeed, their main physiological role is related to the renin-angiotensin-aldosterone system, one of the main physiological pathways for blood pressure regulation and fluid and electrolyte balance [59]. Briefly, the peptide hormone angiotensinogen is secreted by the liver and cleaved by renin to form angiotensin I, which is then converted to angiotensin II by the angiotensin-converting enzyme (ACE), produced by the lungs. Angiotensin II acts on its receptors and modulates several processes, such as aldosterone secretion (in the adrenal glands), water and sodium retention (in the kidneys), sanguine pressure, and vasopressin production (in the CNS) [60]. To treat hypertension, many efforts have been directed towards the creation of drugs acting as ATRs inhibitors. The most famous drug family designed for this purpose is represented by the “Sartans”, which selectively bind to the first isoform of angiotensin receptors [61]. Indeed, ATRs can be divided into four isoforms, AT1, AT2, AT3, and AT4. While the latter two are still in the early stages of research, the first two isoforms have been more deeply characterized. AT1 is mainly found in blood vessels, heart, kidney, brain, and adrenal cortex, mediating vasoconstrictive effects [59]. AT2 receptors are more concentrated in the fetus and neonate, and their functions are more strongly related to neuronal development and excitability [62]. A study by Kawajiri et al. highlighted a reduction in angiotensin II levels in the CSF coming from ALS patients, reporting two opposite consequences: the reduction of protection and repair mediated by AT2, on the one hand, and the reduction of oxidative stress due to AT1 on the other. Indeed, Kawajiri et al. hypothesized that angiotensin II could be downregulated in CSF of ALS patients as a protective reaction, avoiding excessive activation of AT1 [63]. The benefits of AT1 antagonism have also been underlined by Iwasaki et al., who reported evidence of the neurotrophic effects on spinal motor neurons of the drug Olmesartan (Figure 8, panel A), specifically referring to its potential application in ALS [64]. Furthermore, an article by Mammana et al. highlighted the AT1 antagonism-mediated neuroprotective effects of Telmisartan (Figure 8, panel B), also outlining the decrease in neuronal injury and microglial activation caused by it [65].
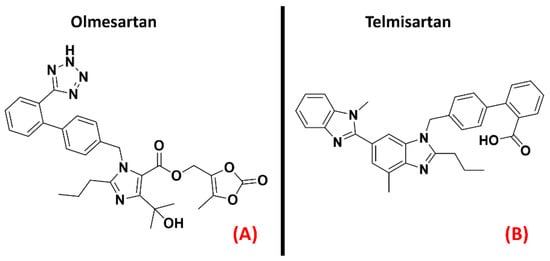
Figure 8. The chemical structures of the AT1 antagonists Olmesartan (A) and Telmisartan (B).
Summing up the information obtainable from the literature, AT1 inhibition could be examined as a potential new therapeutic method of fighting ALS conditions. Both AT1 and AT2 receptors are represented in Figure 9 below.
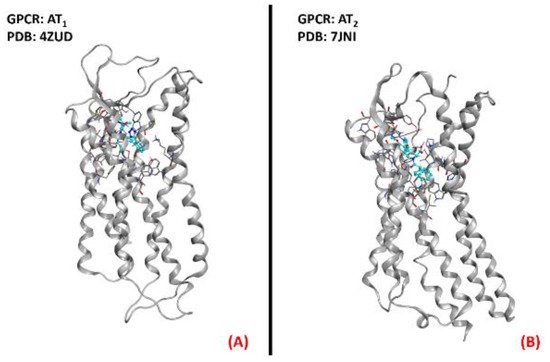
Figure 9. Representation of the three-dimensional structures of the angiotensin II receptors thatcould be considered for ALS treatment. AT1 receptor (A) (sourced from the Protein Data Bank, PDB code: 4ZUD [66], method: X-ray diffraction, resolution: 2.80Å) and AT2 receptor (B) (sourced from the Protein Data Bank, PDB code: 7JNI [67], method: X-ray diffraction, resolution: 3.00 Å). The images were created and rendered with MOE.
2.4. Dopamine Receptors
Dopamine receptors are among the most important and widely studied G-protein coupled receptors, mainly for their important physiological roles in neurotransmission. This class of GPCRs is divided into five isoforms, which are separated into two classes. The first, also called the “D1-like family”, comprises the D1R and D5R receptors, which are coupled to a Gs protein responsible for adenylyl cyclase activation upon binding. The second family, also known as the “D2-like” family, comprises the D2R, D3R, and D4R proteins, all coupled with a Gi protein with inhibitory activity on adenylyl cyclase. Dopamine receptors are localized in different peripheral parts of the organism, such as arteries, heart, and kidneys, but their activities much more determinant within the CNS. Indeed, dopamine is the main neurotransmitter involved in the reward system, and its signaling is of crucial importance for processes such as cognition, memory, and motor control [68]. Dysregulation of the dopaminergic system in the brain represents the main cause of very important diseases such as schizophrenia, attention deficit hyperactivity disorder (ADHD), and Parkinson’s disease [69]. Several articles highlight a correlation between dopamine signaling and ALS development [70]. Researchers have previously reported what was assessed by Liu et al. regarding the protective effect of the A2A receptor on TDP-43 mislocalization [39]. A recent study by Lai et al. details how this beneficial activity can be blocked by D2R activation [71]. Despite this, D2R was also identified as important for the modulation of motor neuron excitability by Huang et al. [72]. Fujimori et al. showed how the treatment with Ropinirole (Figure 10, panel A), an agonist for receptors D2R, D3R, and D4R mainly used for Parkinson’s disease, has neuroprotective effects in ALS models [73]. Additionally, D2R agonists such as Bromocriptine and Sumanirole (both represented in Figure 10, panels B and C, respectively) were tested by Huang et al.,who reported thatt the final effect of such activity on ALS models was an increase in motor neuron survival [74]. Another agonist for the “D2-like family” of dopamine receptors is the R(+) enantiomer of the Parkinson’s disease drug Pramipexole, known as Dexpramipexole (Figure 10, panel D).
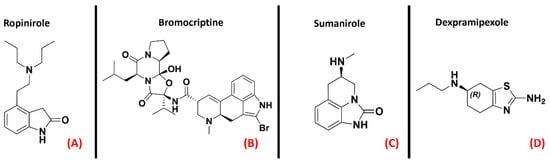
Figure 10. The chemical structures of the dopamine receptor agonists Ropinirole (A), which has an affinity for D2R, D3R, and D4R, Bromocriptine (B), also non-selective with an affinity for D2R, D3R, and D4R, and Sumanirole (C), selective for D2R. (D) Chemical structure of Dexpramipexole (its neuroprotective effects are attributed to dopaminergic-independent activities).
Even if Pramipexole is a powerful agonist of D2R, D3R, and D4R (all depicted in Figure 11) [75], its R(+) enantiomer has a very low affinity for dopamine receptors, so its neuroprotective effects have to be due to a non-dopaminergic action [76]. This molecule has been considered a promising candidate for ALS conditions [77]. After the phase III clinical trial, however, its development in Europe was discontinued [78].
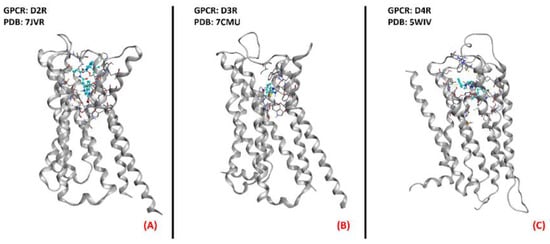
Figure 11. Representation of the three-dimensional structures of the dopamine receptors thatcould be considered for ALS treatment. (A) D2R (sourced the Protein Data Bank, PDB code: 7JVR [79], method: cryo-EM, resolution: 2.80 Å), (B) D3R (sourced from the Protein Data Bank, PDB code: 7CMU [80], method: cryo-EM, resolution: 3.00 Å), and (C) D4R (sourced from the Protein Data Bank, PDB code: 5WIV [81], method: X-ray diffraction, resolution: 2.14 Å). All of the images were created and rendered with MOE.
D2R is still a very relevant target for Parkinson’s disease treatment, but the presented literature concords in considering it also a protein of high therapeutic potential for treating ALS.2.5. Serotonin (5-HT) Receptors
Serotonin (also called 5-hydroxytryptamine, or 5-HT) receptors represent one of the most populated subfamilies of class AGPCRs, consisting of13 G-protein coupled receptor isoforms (5-HT1A, 5-HT1B, 5-HT1D, 5-HT1E, 5-HT1F, 5-HT2A, 5-HT2B, 5-HT2C, 5-HT4, 5-HT5A, 5-HT5B, 5-HT6, and 5-HT7) distributed throughout the entire human organism, and a cation channel (5-HT3), mainly involved in gastrointestinal motility [82].It is also interesting to note that almost all of these isoforms are present in the CNS [83]. Concerning ALS, the serotonin receptor which has gained the greatest popularity is 5-HT2B. An article from Oussini et al., reported that the activity of this biological entity could limit the degeneration of spinal cord mononuclear phagocytes, which is a process typical of neurodegenerative diseases. The ablation of the 5-HT2B gene resulted in an acceleration of ALS progression in mutant SOD1 mouse models. Indeed, they showed that the administration of a 5-HT2B selective antagonist (SB204741, Figure 12, panel A) caused an important reduction in microglia viability, while treatment with the agonist BW723C86 (Figure 12, panel B) induced an increase in viability [84]. Another work by Dentel et al., reported that the spasticity associated with ALS progression could be strongly alleviated by the administration of inverse agonists of 5-HT2B/C such as SB206553and Cyproheptadine (both depicted in Figure 12, panels C and D, respectively) [85]. A recent article by Arnoux et al., on the other hand, highlighted the lack of beneficial effects when ALS-affected SOD1G86R mutants were treated with the 5-HT2B agonist BW723C86 [86]. The main factor that has always limited the development of 5-HT2B agonists is their inherent cardiotoxicity, which can determine valvular heart disease [87]. Two randomized, double-blind, placebo-controlled multicenter studies (phase III) were conducted in 2004 by Meininger et al. to evaluate the potential benefits of Xaliproden (Figure 12, panel E), a 5-HT1A receptor agonist with neuroprotective effects, in ALS patients.
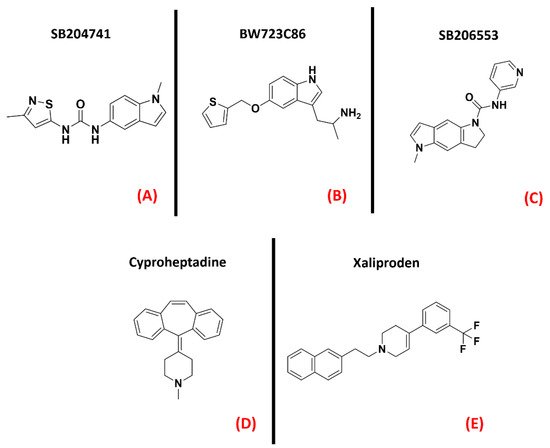
Figure 12. The chemical structures of the serotonin receptor modulators are treated. The selective 5-HT2B antagonist SB204741 (A), the 5-HT2B agonist BW723C86 (B). (C) Structure of the 5-HT2B/C mixed inverse agonistSB206553; (D) chemical structure of Cyproheptadine, a non-selective mixed serotonin receptor antagonist (which is an inverse agonist of 5-HT2B). (E) Structureof the 5-HT1A receptor agonist Xaliproden.
Despite the promising outcomes of the prior experiments [88], no effective slowing down in the progression of the pathology was evidenced. Even if some limitations have been encountered in serotoninergic modulation for motor neuron disease, the 5-HT receptors have been demonstrated to be targets of relevance in the ALS scenario. The three-dimensional structures of 5-HT1A, 5-HT2B, and 5-HT2C are represented in Figure 13.
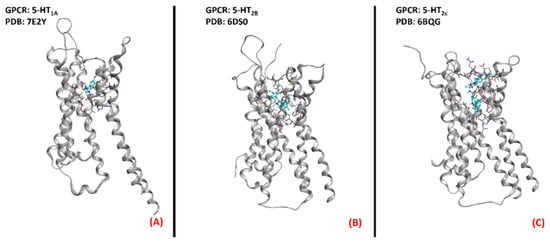
Figure 13. Three-dimensional structures of the serotonin receptors that could be considered for ALS treatment. (A) 5-HT1A (sourced from the Protein Data Bank, PDB code: 7E2Y [89], method: cryo-EM, resolution: 3.00 Å), (B) 5-HT2B (sourced from the Protein Data Bank, PDB code: 6DS0 [90], method: X-ray diffraction, resolution: 3.19 Å), and (C) 5-HT2C (sourced from the Protein Data Bank, PDB code: 6BQG [91], method: X-ray diffraction, resolution: 3.00 Å). All of the images were created and rendered with MOE.
2.6. GPR17 Receptor
GPR17 (also known as “uracil nucleotide/cysteinyl leukotriene receptor”) is a protein belonging to the 15th subfamily of classA GPCRs. One of its peculiarities is that its structure is phylogenetically related to both cysteinyl leukotriene (CysLT) receptors and to purinergic P2Y receptors [92]. This receptor is activated by uracil nucleotides such as UDP, UDP-glucose, and UDP-galactose, but is also sensitive to CysLTs, like Leukotriene D4 and C4 [93]. GPR17 is mainly expressed in the CNS (but also in kidneys, heart, and generally in organs that can experience ischemic damage), and a more pronounced presence of this protein has been highlighted in oligodendrocyte precursor cells (OPCs). Upregulation of GPR17 can be observed in neuronal cells surrounding an ischemic-injured area, making this protein a marker for cellular stress and death.It has been reported in the literature that in the case of a demyelinating event, GPR17 is involved in the remyelination process, but the mechanism of its involvement is still debated [94]. What is known is thatGPR17 is deputed to accompany the OPCs in the early stages of their differentiation process, and so its downregulation is necessary for these cells to complete their maturation. As a result of this, overexpression of this protein leads to incomplete OPC development, impairing myelination and promoting inflammatory responses [95]. Moreover, GPR17 upregulation in neurons was linked to increased cell damage by Zhao et al., who also observed that its knockdown attenuated neuronal injury and microgliosis [96]. In the field of ALS, GPR17 was demonstrated by Bonfanti et al., to be upregulated in the spinal cord of SOD1G93A mouse models [97]. As reported in a recent study by Raffaele et al., while the application of non-selective GPR17 antagonists such as HAMI3379 (Figure 14, panel A) or Montelukast (a marketed CysLT receptor inhibitor, which is represented in Figure 14, panel B) has been shown to improve remyelination processes (as also demonstrated by Merten et al. for the first of these two molecules [98]), the implementation of agonists has also been demonstrated to be beneficial in pushing OPCs to start differentiating [99]. Jin et al., asserted that the inhibition of GPR17 by Cangrelor (Figure 14, panel C) results in the amelioration of cognitive deficits through the inhibition of oxidative stress and neuroinflammation in Alzheimer’s Disease mouse models [100].
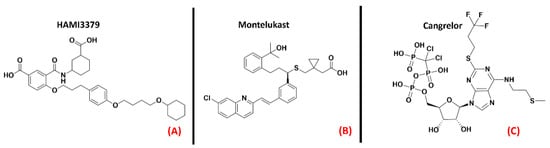
Figure 14. The chemical structures of the GPR17 inhibitors. The non-selective inhibitors HAMI3397, Montelukast (sold as a CysLT receptors inhibitor for asthma), and Cangrelor (an antiplatelet drug, reversible inhibitor of P2Y12 receptor), respectively (A–C).
Marschallinger et al., in a recent paper, highlighted a restoration in cognitive function and a reduction in neuroinflammation in rats treated with Montelukast [101]. Another study by Burnstock et al., indicated that the in vivo knockdown of GPR17 markedly reduced brain damage [102]. The information available nowadays converges in indicating GPR17 (which three-dimensional structure is provided in Figure 15) as a promising target for neuroinflammation and neurodegeneration diseases. Even if, at the present moment, these efforts are more focused on multiple sclerosis treatment, GPR17 regulation for ALS is also attracting increasing interest from the scientific community.
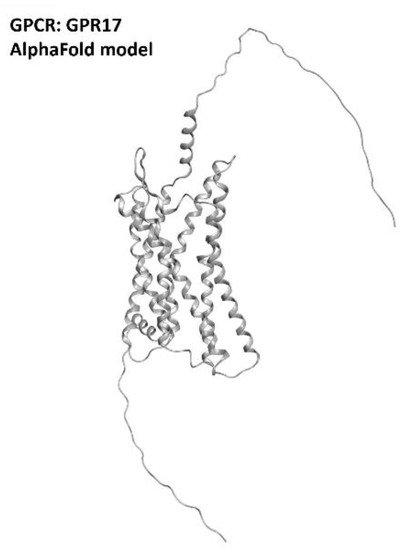
Figure 15. Structure of the GPR17 receptors (with no experimentally resolved structure available, the model sourced from the AlphaFold database is presented). The image was created and rendered with MOE.
2.7. Adrenergic Receptor β2
Adrenergic receptors are part of the 17th subfamily of class A GPCRs and are divided into nine different isoforms (α1A, α1B, α1D, α2A, α2B, α2C, β1, β2, β3), which are involved in very important physiological functions such as smooth muscle contraction and relaxation, heart muscle contraction (mainly receptors β1 and β2) [103], and glycogenolysis [104]. The research to find a link between adrenergic transmission and ALS has been focused on the β2 isoforms of this GPCR. Historically, β2 agonists represent one of the main solutions for asthma therapy [105], and drugs such as Salbutamol, Clenbuterol, and Formoterol (all depicted in Figure 16) are an example of this. A recent work by Bartus et al. highlighted the potentialities of β2 agonists for ALS, reporting that the downstream effects of these molecules can be useful for protecting spinal cord neurons, both preserving and/or restoring their function [106].
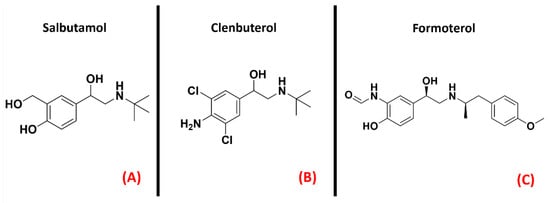
Figure 16. The chemical structures of the adrenergic β2 receptor agonists Salbutamol (A), Clenbuterol (B), and Formoterol (C).
The pathways responsible for such outcomes presented by Bartus et al. are very biologically important for cell homeostasis, such as the cAMP/PKA/CREB pathway, the PI3K-Akt-mTOR pathway, and the PKA/SIRT1 pathway. Other than neuroprotection, other effects attributed by the researchers to this class of ligands are the increase in muscle strength and the amelioration of mitochondrial function. Another study by Teng et al. reported a favorable effect of the β2 agonist Clenbuterol on SOD1G93A mice [107]. The outcomes of these studies open new possibilities in drug discovery for ALS, focusing special attention on adrenergic β2 receptor modulation. A three-dimensional representation of the adrenergic β2 receptor is provided in Figure 17.
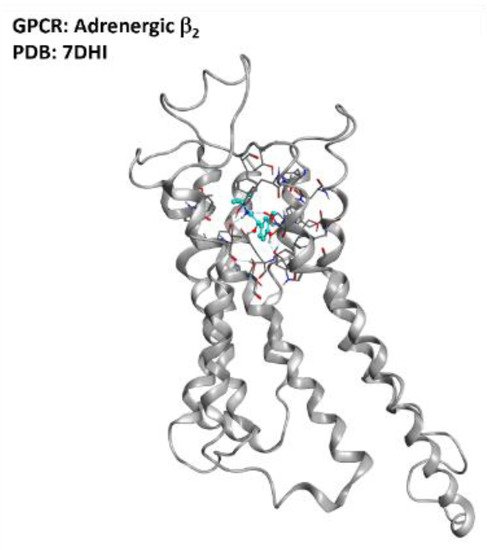
Figure 17. Representation of the structure of the adrenergic β2 receptor (sourced from the Protein Data Bank, PDB code: 7DHI [108], method: cryo-EM, resolution: 3.26 Å). The image was created and rendered with MOE.
2.8. Histamine Receptors
Histamine receptors represent a group of class A GPCRs that has attracted a lot of interest in the pharmaceutical world in recent decades. This family is composed of four different members (H1, H2, H3, and H4), each with a specific localization in the organism. Their functions, vary from one isoform to another, ranging from vasoconstriction (H1) to gastric acid secretion (H2), to neurotransmitter release (H3), to immunoregulation (mainly H2 and H4) [109]. For each histamine receptor, the research has mainly focused on the mechanism of antagonism, of which several marketed drugs are still now relevant examples (e.g., Cetirizine, Figure 18, panel A, for H1 antagonism; or Famotidine, represented in Figure 18, panel B, for H2 blockage) [110].
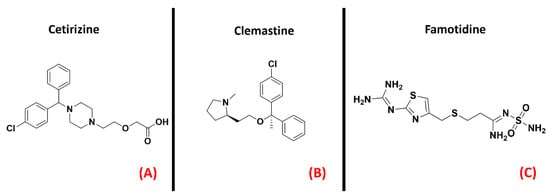
Figure 18. The chemical structures of the H1 receptor antagonists Cetirizine (A) and Clemastine (B). The H2 receptor antagonist Famotidine is also reported (C).
An article from Apolloni et al. reported the involvement of histaminergic signals in ALS progression, highlighting that histamine receptors are dysregulated in the cortex, spinal cord, and hypothalamus of SOD1G93A ALS-affected mice [111]. Histamine could counteract the pro-inflammatory phenotype of microglia, mainly through its H1 and H4 receptor isoforms. This would be mediated by both the reduction of NOX-2 and NF-kB expression and the increase in production of other species, such as IL-6 and IL-10. Another work by Volontè et al. highlighted the neuroprotective effects of histamine signaling in ALS, again giving higher relevance to H1R and H4R [112]. On the other hand, Zhang et al. described H1 and H4 receptors as being responsible for pro-inflammatory cytokine release in microglia, while H2 and H3 were considered to be the main actors of anti-inflammation in that environment (the H2 receptor is depicted in its three-dimensional structure in Figure 19, panel B) [113]. Another article by Apolloni et al. reported an amelioration in ALS progression of SOD1G93A mice treated with the anti-histaminergic drug Clemastine [114]. Even if much more remains to be understood about the specific role of each histamine receptor isoform in ALS progression, what is certain is that this GPCR family has already proven to be a promising target for drug development against neuroinflammation and neurodegeneration.
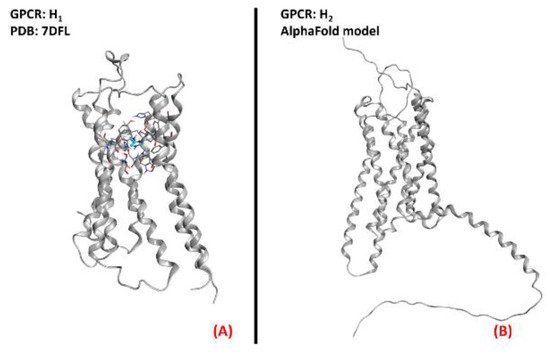
Figure 19. Representation of the three-dimensional structures of the histamine receptors that could be considered for ALS treatment. (A) The H1 receptor (sourced from the Protein Data Bank, PDB code: 7DFL [115], method: cryo-EM, resolution: 3.30 Å) and (B) the H2 receptor (with no experimentally resolved structure available, the model sourced from the AlphaFold [116] database is presented).The images were created and rendered with MOE. The H3 and H4 receptors both lack experimentally resolved structures, but their AlphaFold models are available.
2.9. Cannabinoid Receptors
A group of class AGPCRs that are of high interest at the present date is certainly the cannabinoid receptors. These biological entities are the main actors in the endocannabinoid system, and play relevant roles in several physiological processes. Indeed, the first of its two main isoforms, called CB1, is mainly located in both the central and peripheral nervous system, acting as a neurotransmitter release modulator in response to the binding of its agonists (mainly anandamide, but also 2-arachidonoylglycerol, both represented in Figure 20, panels A and B, respectively). In the majority of cases, CB1 is coupled with Gi/o protein, leading to adenylyl cyclase inhibition and consequent decrease in cAMP upon activation. The final effect of such an action is the reduction of neurotransmitter release in the synapse. On the other hand, the CB2 receptor is mainly localized on the surface of the immune system cells. Its main agonist is 2-arachidonoylgycerol, binding of which leads to the inhibition of adenylyl cyclase through Gi/o subunit action [117]. The final main effect is immunosuppression [118]. As reported by an article from Giacoppo and Mazzon, several studies have shown how the application of cannabinoid receptor agonists in SOD1G93A mouse models of ALS could be beneficial for the neuroprotective effects mediated by them [119]. Similarly, in 2019,Urbi et al. performed a meta-analysis on the studies regarding the application of cannabinoids in ALS murine models, highlighting the effective concordance in assessing that their application leads to a delay in disease progression [120]. A study by Shoemaker et al. highlighted that the increase in survival could be more addressable to the CB2 isoform, showing that the administration of the CB2 selective agonist AM-1241 (Figure 20, panel C) increased survival by 56% [121]. This molecule was also examined for cannabinoid-mediated ALS treatment by Kim et al., with similar results [122].
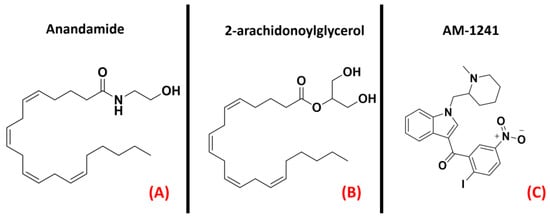
Figure 20. The chemical structures of the endogenous CB1 and CB2 receptors agonists anandamide (A) and 2-arachidonoylglycerol (B). The selective CB2 receptor agonist AM-1241 is also reported (C).
In addition to this, Bilsland et al. reported that the knock-out of CB1 receptors in SOD1G39A ALS-affected mice had no appreciable effect on disease onset [123], and regarding this, Shoemaker et al. reported that the activation of CB1 could exacerbate disease progression [121]. The literature available today regarding the application of molecules acting on the endocannabinoid system for ALS treatment converges in the possible evaluation of a therapy based on CB2 selective agonists. A three-dimensional representation of both CB1 and CB2 receptorsis provided in Figure 21.
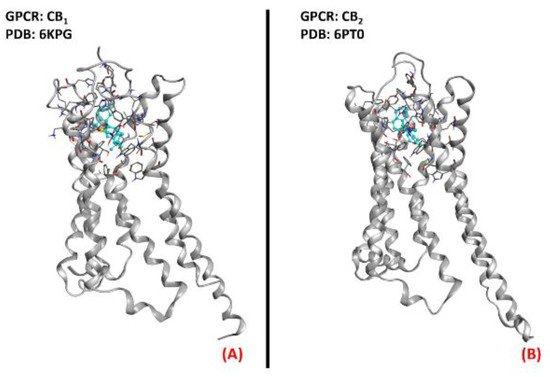
Figure 21. The structures of (A) the CB1 receptor (sourced from the Protein Data Bank, PDB code: 6KPG [124], method: cryo-EM, resolution: 3.00 Å) and (B) the CB2 receptor (sourced from the Protein Data Bank, PDB code: 6PT0 [125], method: cryo-EM, resolution: 3.20 Å). The images were created and rendered with MOE.
2.10. Prostaglandin E2 Receptor (PGE2R)
Prostaglandin E2 receptors (PGE2) are a series of class A GPCRs that selectively bind to prostaglandin E2 (also known as dinoprostone), an endogenous arachidonic acid derivative of high importance for several physiological functions. PGE2 can be divided into four isoforms, named E1, E2, E3, and E4 (all represented in their three-dimensional structure in Figure 22). With the exception of the first, which stimulates phospholipase C if agonized, the other isoforms act on adenylyl cyclase and, specifically, the EP2 and EP4 isoforms (coupled with a Gs subunit) stimulate its function when agonized, while EP3 inhibits AC through its action (being coupled to a Gi/o subunit) [126]. EP1 function has been correlated with hyperalgesia [127], immunoregulation [128], and colon cancer progression [129]. EP2, which is active in the reproductive, visual, cardiovascular, skeletal, and nervous systems, has also been strictly related to tumor promotion, as highlighted in a 2018 study bySun and Li [130]. Minor correlations with cancer have been reported for EP3, which is also important for a large variety of functions, ranging from digestion [131] to blood pressure [132] and clotting [133], in addition to pain management [134]. The spectrum of systems in which the fourth isoform of PGE2 receptors, EP4, is involved is also very wide. Additionally, in this case, EP4 has been reported to be hyper-expressed in various types of cancer, mainly prostate cancer [135]. Talking about ALS onset and progression, Iłzecka found increased levels of PGE2 in the cerebrospinal fluid of ALS patients, and therefore concluded that this mediator could play a role in disease progression, suggesting that its inhibition could be beneficial [136]. Additionally, Kosuge et al., highlighted the role of PGE2 in the ROS generation pathway, focusing on its impact on ALS conditions. The same conclusion was reported in 2008 by Liang et al., who suggested EP2 inhibition as a novel way to treat the neuroinflammation typical in ALS [137]. On the other hand, PGE2 receptors were proposed to have an unexpected neuroprotective effect on motor neurons by Bilak et al., who reported that the neuroinflammatory process typical of ALS was mainly due to COX-2-mediated, prostaglandin-independent processes [138]. Taken together, all of these studies converge in evaluating PGE2 receptors as interesting pharmacological targets for ALS, being strongly correlated with the significant neuroinflammation characterizing the pathology.
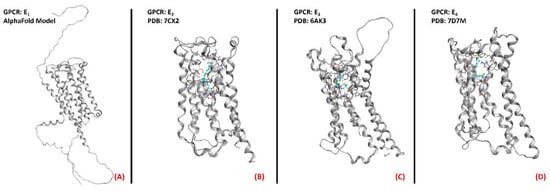
Figure 22. Structure of the PGE2 receptors (A) EP1 (with no experimentally resolved structure available, the model sourced from the AlphaFold database is presented), (B) EP2 (sourced from the Protein Data Bank, PDB code: 7CX2 [139], method: cryo-EM, resolution: 2.80 Å), (C) EP3 (sourced from the Protein Data Bank, PDB code: 6AK3 [140], method: X-ray diffraction, resolution: 2.90 Å), and (D) EP4 (sourced from the Protein Data Bank, PDB code: 7D7M [141], method: cryo-EM, resolution: 3.30 Å). All of the images were created and rendered with MOE.
2.11. Vasoactive Intestinal Peptide Receptors
The receptors for the vasoactive intestinal peptide are part of the first subfamily of class B GPCRs. As part of this group of proteins, these receptors are responsive to signals mediated by the peptide hormone VIP (vasoactive intestinal polypeptide), formed by 28 amino acids and belonging to the glucagon/secretin superfamily [142]. After being produced by organs such as the gut, pancreas, and brain, VIP act in different physiological functions depending on the target tissue and the receptor isoform interacting with it. Indeed, two vasoactive intestinal polypeptide receptor isoforms are known, namely VPAC1 and VPAC2 (both represented in Figure 23). Both of these proteins are highly expressed throughout the human body, from the smooth muscle of the GI tract and blood vessels to the reproductive system, lungs, spleen, and brain [143]. Their activity is mediated by a Gs protein, and involves the activation of adenylyl cyclase upon binding to VIP, consequently activating the protein kinase A (PKA) [144].
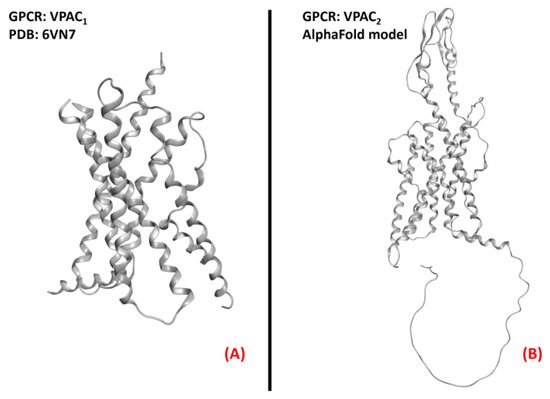
Figure 23. Structure of the receptors (A) VPAC1 (sourced from the Protein Data Bank, PDB code: 6VN7 [145], method: cryo-EM, resolution: 3.20 Å), and (B) VPAC2 (with no experimentally resolved structure available, the model sourced from the AlphaFold database is presented). The images were created and rendered with MOE.
The implication of VIP in the CNS has been noticed when studying both the circadian rhythm and schizophrenia [146], but its importance as a potential target for ALS therapy is of more recent discovery. In 2008, Staines considered the possibility of studying vasoactive neuropeptides for degenerating pathologies such as MS and ALS [147], and a previous article by Iwasaki et al. specifically referred to the neurotrophic properties exerted by VIP in the degenerating diseases of motor neurons [148]. Solés-Tarrés et al. analyzed the neuroprotective effects of both VIP and PACAP (pituitary adenylate cyclase-activating polypeptide, a peptide hormone binding to both VPACs and PACAP receptors), also evaluating the synthetic derivatives available to mimic their action, with a special focus on the intrinsic pharmacokinetic problems of these species [149]. Waschek already identified both VIP and PACAP as promising targets for neuroinflammation in the CNS [150], as pointed out again in recent work by Martinez et al. [151]. The interaction with receptors of vasoactive peptides has been demonstrated to be a promising way to counteract the neuroinflammatory and degenerative effects of ALS, mainly through biological and/or chemical species mimicking the functions of the endogenous peptides. Further research on this topic will define the best way to accomplish this task.
2.12. Metabotropic Glutamate Receptors (mGluRs)
Metabotropic glutamate receptors (mGluRs) are GPCRs belongingto class C of this family of proteins. As the name suggests, these entities bind to the neurotransmitter glutamate, exerting different functions in both the central and peripheral nervous systems. They can be divided into threegroups, with the first beingcomposed of mGluR1 and mGluR5, predominantly postsynaptic, which, once activated, cause the stimulation of phospholipase C (PLC) through Gq-mediated signaling. Group II (formed by mGluRs 2 and 3) and III (of which mGluRs 4, 6, 7, and 8 are a part) receptors are mainly presynaptic and are all coupled with a Gi/0 subunit, which inhibits the activation of adenylyl cyclase, causing presynaptic inhibition. The functions of this family of proteins are majorly related to the nervous system, from the modulation of neurotransmission (e.g., gabaergic and dopaminergic) and of other proteins’ signaling (e.g., NMDA receptors), to synaptic plasticity regulation [152]. This being said, it appears clear that the possibility of their involvement in ALS onset and progression is more than possible. Anneser et al. found a strong upregulation of mGluRs in the spinal cord with ALS, leading to the propagation of glial proliferation [153]. Hyperactivity of group I mGluRs has been correlated with neuroinflammation. Indeed, as demonstrated by Milanese et al., SOD1G93A ALS-affected mice with mGluR1 knockdown experience a reduction in microglia and astrocyte activation, decreasing mitochondrial damage and improving survival [154], and this phenomenon was also highlighted by Rossi et al. [155]. Anneser et al. showed the beneficial and protective effects of both agonism and antagonism of group I mGluRs for motor neuron disease, while less promising effects were derived from modulation of other mGluRs [156]. Crupi et al. recently pointed out that the beneficial therapeutic modulation of mGluRs is usually achieved through the reduction of the excitotoxicity drive via mGluR I inhibition or mGluR II and III agonism [157]. In conclusion, the literature supports the possibility of investing resources in the treatment of motor neuron diseases via mGluR modulation. A three-dimensional representation of mGluR1, mGluR2, and mGluR4 receptorsis provided in Figure 24.
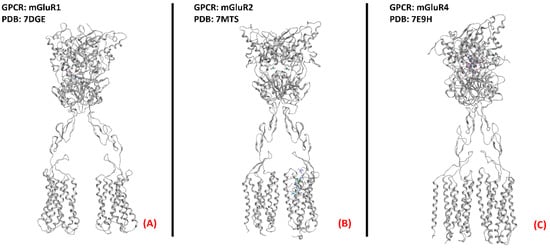
Figure 24. One example of each group of the metabotropic glutamate receptors. (A) mGluR1, a member of the first group of mGluRs (sourced from the Protein Data Bank, PDB code: 7DGE [158], method: cryo-EM, resolution: 3.65 Å), (B) mGluR2 (owing to mGluRs group II, sourced from the Protein Data Bank, PDB code: 7MTS [159], method: cryo-EM, resolution: 3.20 Å), and (C) mGluR4, part of group III of the mGluRs (sourced from the Protein Data Bank, PDB code: 7E9H [160], method: cryo-EM, resolution: 4.00 Å). All of the images were created and rendered with MOE.
References
- Adams, J.; Lee, M.; Peng, W. Critical Review of Complementary and Alternative Medicine Use in Amyotrophic Lateral Sclerosis: Prevalence and Users’ Profile, Decision-Making, Information Seeking, and Disclosure in the Face of a Lack of Efficacy. Neurodegener. Dis. 2018, 18, 225–232.
- Heckman, C.; Enoka, R.M. Physiology of the motor neuron and the motor unit. In Handbook of Clinical Neurophysiology; Elsevier: Amsterdam, The Netherlands, 2004; pp. 119–147.
- Wijesekera, L.C.; Leigh, P.N. Amyotrophic lateral sclerosis. Orphanet J. Rare Dis. 2009, 4, 3.
- Hardiman, O.; Al-Chalabi, A.; Chiò, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Prim. 2017, 3, 17071.
- Ortí, J.D.L.R.; Armero, J.; Sanchis-Sanchis, C.; Sancho-Castillo, S.; Salazar, A.; Caplliure-Llopis, J.; Navarro-Illana, E.; Barrios, C.; Escribá-Alepuz, J.; Benlloch, M. Muscle Function Differences between Patients with Bulbar and Spinal Onset Amyotrophic Lateral Sclerosis. Does It Depend on Peripheral Glucose? J. Clin. Med. 2021, 10, 1582.
- Van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; van den Berg, L.H. Amyotrophic lateral sclerosis. Lancet 2017, 390, 2084–2098.
- National Institute of Neurological Disorders and Stroke. Amyotrophic Lateral Sclerosis (ALS) Fact Sheet.
- Saitoh, Y.; Takahashi, Y. Riluzole for the treatment of amyotrophic lateral sclerosis. Neurodegener. Dis. Manag. 2020, 10, 343–355.
- Breiner, A.; Zinman, L.; Bourque, P.R. Edaravone for amyotrophic lateral sclerosis: Barriers to access and lifeboat ethics. Can. Med. Assoc. J. 2020, 192, E319–E320.
- Jaiswal, M.K. Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med. Res. Rev. 2018, 39, 733–748.
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929
- Boddy, S.L.; Giovannelli, I.; Sassani, M.; Cooper-Knock, J.; Snyder, M.P.; Segal, E.; Elinav, E.; Barker, L.A.; Shaw, P.J.; McDermott, C.J. The gut microbiome: A key player in the complexity of amyotrophic lateral sclerosis (ALS). BMC Med. 2021, 19, 13.
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133.
- Jo, M.; Lee, S.; Jeon, Y.-M.; Kim, S.; Kwon, Y.; Kim, H.-J. The role of TDP-43 propagation in neurodegenerative diseases: Integrating insights from clinical and experimental studies. Exp. Mol. Med. 2020, 52, 1652–1662.
- Suk, T.; Rousseaux, M.W.C. The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol. Neurodegener. 2020, 15, 45.
- Berdyński, M.; Miszta, P.; Safranow, K.; Andersen, P.M.; Morita, M.; Filipek, S.; Żekanowski, C.; Kuźma-Kozakiewicz, M. SOD1 mutations associated with amyotrophic lateral sclerosis analysis of variant severity. Sci. Rep. 2022, 12, 103.
- Ishigaki, S.; Sobue, G. Importance of Functional Loss of FUS in FTLD/ALS. Front. Mol. Biosci. 2018, 5, 44
- Eck, R.J.; Kraemer, B.C.; Liachko, N.F. Regulation of TDP-43 phosphorylation in aging and disease. GeroScience 2021, 43, 1605–1614.
- Guo, W.; Vandoorne, T.; Steyaert, J.; Staats, K.A.; Van Den Bosch, L. The multifaceted role of kinases in amyotrophic lateral sclerosis: Genetic, pathological and therapeutic implications. Brain 2020, 143, 1651–1673.
- Palomo, V.; Nozal, V.; Rojas-Prats, E.; Gil, C.; Martinez, A. Protein kinase inhibitors for amyotrophic lateral sclerosis therapy. J. Cereb. Blood Flow Metab. 2020, 178, 1316–1335.
- Miller, R.G.; Mitchell, J.D.; Moore, D.H. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst. Rev. 2012, 65, CD001447.
- Moro, S.; Bissaro, M. Rethinking to riluzole mechanism of action: The molecular link among protein kinase CK1δ activity, TDP-43 phosphorylation, and amyotrophic lateral sclerosis pharmacological treatment. Neural Regen. Res. 2019, 14, 2083–2085.
- Corcia, P.; Beltran, S.; Bakkouche, S.; Couratier, P. Therapeutic news in ALS. Rev. Neurol. 2021, 177, 544–549.
- Springer Nature. AdisInsight—Small-Molecules in Clinical Trials for ALS.
- Chen, H.; Kankel, M.W.; Su, S.C.; Han, S.W.S.; Ofengeim, D. Exploring the genetics and non-cell autonomous mechanisms underlying ALS/FTLD. Cell Death Differ. 2018, 25, 648–662.
- Insel, P.A.; Wilderman, A.; Zambon, A.C.; Snead, A.N.; Murray, F.; Aroonsakool, N.; McDonald, D.S.; Zhou, S.; McCann, T.; Zhang, L.; et al. G Protein–Coupled Receptor (GPCR) Expression in Native Cells: “Novel” endoGPCRs as Physiologic Regulators and Therapeutic Targets. Mol. Pharmacol. 2015, 88, 181–187.
- Gacasan, S.B.; Baker, D.L.; Parrill, A.L. G protein-coupled receptors: The evolution of structural insight. AIMS Biophys. 2017, 4, 491–527.
- Lee, Y.; Basith, S.; Choi, S. Recent Advances in Structure-Based Drug Design Targeting Class A G Protein-Coupled Receptors Utilizing Crystal Structures and Computational Simulations. J. Med. Chem. 2017, 61, 1–46.
- Heng, B.C.; Aubel, D.; Fussenegger, M. An overview of the diverse roles of G-protein coupled receptors (GPCRs) in the pathophysiology of various human diseases. Biotechnol. Adv. 2013, 31, 1676–1694.
- Zarrinmayeh, H.; Territo, P.R. Purinergic Receptors of the Central Nervous System: Biology, PET Ligands, and Their Applications. Mol. Imaging 2020, 19, 1536012120927609.
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of Adenosine Receptors: The State of the Art. Physiol. Rev. 2018, 98, 1591–1625.
- Gomes, C.; Kaster, M.; Tome, A.; Agostinho, P.; Cunha, R.A. Adenosine receptors and brain diseases: Neuroprotection and neurodegeneration. Biochim. Biophys. Acta 2011, 1808, 1380–1399.
- Vincenzi, F.; Corciulo, C.; Targa, M.; Casetta, I.; Gentile, M.; Granieri, E.; Borea, P.A.; Popoli, P.; Varani, K. A2Aadenosine receptors are up-regulated in lymphocytes from amyotrophic lateral sclerosis patients. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 406–413.
- Yoshida, Y.; Une, F.; Utatsu, Y.; Nomoto, M.; Furukawa, Y.; Maruyama, Y.; Machigashira, N.; Matsuzaki, T.; Osame, M. Adenosine and Neopterin Levels in Cerebrospinal Fluid of Patients with Neurological Disorders. Intern. Med. 1999, 38, 133–139.
- Potenza, R.L.; Armida, M.; Ferrante, A.; Pèzzola, A.; Matteucci, A.; Puopolo, M.; Popoli, P. Effects of chronic caffeine intake in a mouse model of amyotrophic lateral sclerosis. J. Neurosci. Res. 2013, 91, 585–592.
- Mori, A.; Cross, B.; Uchida, S.; Walker, J.K.; Ristuccia, R. How Are Adenosine and Adenosine A2A Receptors Involved in the Pathophysiology of Amyotrophic Lateral Sclerosis? Biomedicines 2021, 9, 1027.
- Ng, S.K.; Higashimori, H.; Tolman, M.; Yang, Y. Suppression of adenosine 2a receptor (A2aR)-mediated adenosine signaling improves disease phenotypes in a mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2015, 267, 115–122.
- Mojsilovic-Petrovic, J.; Jeong, G.-B.; Crocker, A.; Arneja, A.; David, S.; Russell, D.; Kalb, R.G. Protecting Motor Neurons from Toxic Insult by Antagonism of Adenosine A2a and Trk Receptors. J. Neurosci. 2006, 26, 9250–9263.
- Liu, Y.-J.; Ju, T.; Chen, H.-M.; Jang, Y.-S.; Lee, L.-M.; Lai, H.-L.; Tai, H.-C.; Fang, J.-M.; Lin, Y.-L.; Tu, P.-H.; et al. Activation of AMP-activated protein kinase α1 mediates mislocalization of TDP-43 in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2014, 24, 787–801.
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242.
- Segala, E.; Guo, D.; Cheng, R.K.Y.; Bortolato, A.; Deflorian, F.; Doré, A.S.; Errey, J.C.; Heitman, L.H.; Ijzerman, A.P.; Marshall, F.H.; et al. Controlling the Dissociation of Ligands from the Adenosine A2A Receptor through Modulation of Salt Bridge Strength. J. Med. Chem. 2016, 59, 6470–6479.
- Chemical Computing Group ULC. Molecular Operating Environment (MOE); Chemical Computing Group ULC: Montreal, QC, Canada, 2021.
- Burnstock, G. Purinergic Signalling: Therapeutic Developments. Front. Pharmacol. 2017, 8, 661.
- Morillas, A.G.; Besson, V.; Lerouet, D. Microglia and Neuroinflammation: What Place for P2RY12? Int. J. Mol. Sci. 2021, 22, 1636.
- Amadio, S.; Parisi, C.; Montilli, C.; Carrubba, A.S.; Apolloni, S.; Volonté, C. P2Y12 Receptor on the Verge of a Neuroinflammatory Breakdown. Mediat. Inflamm. 2014, 2014, 975849.
- Jacobson, K.A.; Delicado, E.G.; Gachet, C.; Kennedy, C.; Von Kügelgen, I.; Li, B.; Miras-Portugal, M.T.; Novak, I.; Schöneberg, T.; Perez-Sen, R.; et al. Update of P2Y receptor pharmacology: IUPHAR Review 27. J. Cereb. Blood Flow Metab. 2020, 177, 2413–2433.
- D’Ambrosi, N.; Finocchi, P.; Apolloni, S.; Cozzolino, M.; Ferri, A.; Padovano, V.; Pietrini, G.; Carrì, M.T.; Volonté, C. The Proinflammatory Action of Microglial P2 Receptors Is Enhanced in SOD1 Models for Amyotrophic Lateral Sclerosis. J. Immunol. 2009, 183, 4648–4656.
- Kobayashi, K.; Yamanaka, H.; Fukuoka, T.; Dai, Y.; Obata, K.; Noguchi, K. P2Y12 Receptor Upregulation in Activated Microglia Is a Gateway of p38 Signaling and Neuropathic Pain. J. Neurosci. 2008, 28, 2892–2902.
- Moore, C.S.; Ase, A.R.; Kinsara, A.; Rao, V.T.; Michell-Robinson, M.; Leong, S.Y.; Butovsky, O.; Ludwin, S.K.; Séguéla, P.; Bar-Or, A.; et al. P2Y12 expression and function in alternatively activated human microglia. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e80.
- Hughes, C.E.; Nibbs, R.J.B. A guide to chemokines and their receptors. FEBS J. 2018, 285, 2944–2971.
- Agle, K.A.; Vongsa, R.A.; Dwinell, M.B. Calcium Mobilization Triggered by the Chemokine CXCL12 Regulates Migration in Wounded Intestinal Epithelial Monolayers. J. Biol. Chem. 2010, 285, 16066–16075.
- La Cognata, V.; Golini, E.; Iemmolo, R.; Balletta, S.; Morello, G.; De Rosa, C.; Villari, A.; Marinelli, S.; Vacca, V.; Bonaventura, G.; et al. CXCR2 increases in ALS cortical neurons and its inhibition prevents motor neuron degeneration in vitro and improves neuromuscular function in SOD1G93A mice. Neurobiol. Dis. 2021, 160, 105538.
- Rabinovich-Nikitin, I.; Ezra, A.; Barbiro, B.; Rabinovich-Toidman, P.; Solomon, B. Chronic administration of AMD3100 increases survival and alleviates pathology in SOD1G93A mice model of ALS. J. Neuroinflamm. 2016, 13, 123.
- Liu, E.; Karpf, L.; Bohl, D. Neuroinflammation in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia and the Interest of Induced Pluripotent Stem Cells to Study Immune Cells Interactions with Neurons. Front. Mol. Neurosci. 2021, 14, 767041.
- Perner, C.; Perner, F.; Stubendorff, B.; Förster, M.; Witte, O.W.; Heidel, F.H.; Prell, T.; Grosskreutz, J. Dysregulation of chemokine receptor expression and function in leukocytes from ALS patients. J. Neuroinflamm. 2018, 15, 99.
- Liu, K.; Wu, L.; Yuan, S.; Wu, M.; Xu, Y.; Sun, Q.; Li, S.; Zhao, S.; Hua, T.; Liu, Z.-J. Structural basis of CXC chemokine receptor 2 activation and signalling. Nature 2020, 585, 135–140.
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589, PMCID:PMC8371605.
- Wu, B.; Chien, E.Y.T.; Mol, C.D.; Fenalti, G.; Liu, W.; Katritch, V.; Abagyan, R.; Brooun, A.; Wells, P.; Bi, F.C.; et al. Structures of the CXCR4 Chemokine GPCR with Small-Molecule and Cyclic Peptide Antagonists. Science 2010, 330, 1066–1071.
- Singh, K.D.; Karnik, S.S. Angiotensin Receptors: Structure, Function, Signaling and Clinical Applications. J. Cell Signal. 2016, 1, 111.
- Ames, M.K.; Atkins, C.E.; Pitt, B. The renin-angiotensin-aldosterone system and its suppression. J. Veter. Intern. Med. 2018, 33, 363–382.
- Miura, S.-I.; Karnik, S.S.; Saku, K. Review: Angiotensin II type 1 receptor blockers: Class effects versus molecular effects. J. Renin-Angiotensin-Aldosterone Syst. 2010, 12, 1–7.
- Akishita, M.; Ito, M.; Lehtonen, J.Y.; Daviet, L.; Dzau, V.J.; Horiuchi, M. Expression of the AT2 receptor developmentally programs extracellular signal-regulated kinase activity and influences fetal vascular growth. J. Clin. Investig. 1999, 103, 63–71.
- Kawajiri, M.; Mogi, M.; Higaki, N.; Tateishi, T.; Ohyagi, Y.; Horiuchi, M.; Miki, T.; Kira, J.-I. Reduced angiotensin II levels in the cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Acta Neurol. Scand. 2009, 119, 341–344.
- Iwasaki, Y.; Ichikawa, Y.; Igarashi, O.; Kinoshita, M.; Ikeda, K. Trophic effect of Olmesartan, a novel AT1R antagonist, on spinal motor neuronsin vitroandin vivo. Neurol. Res. 2002, 24, 468–472.
- Mammana, S.; Fagone, P.; Cavalli, E.; Basile, M.S.; Petralia, M.C.; Nicoletti, F.; Bramanti, P.; Mazzon, E. The Role of Macrophages in Neuroinflammatory and Neurodegenerative Pathways of Alzheimer’s Disease, Amyotrophic Lateral Sclerosis, and Multiple Sclerosis: Pathogenetic Cellular Effectors and Potential Therapeutic Targets. Int. J. Mol. Sci. 2018, 19, 831.
- Zhang, H.; Unal, H.; Desnoyer, R.; Han, G.W.; Patel, N.; Katritch, V.; Karnik, S.S.; Cherezov, V.; Stevens, R.C. Structural Basis for Ligand Recognition and Functional Selectivity at Angiotensin Receptor. J. Biol. Chem. 2015, 290, 29127–29139.
- Perryman, R. Inhibition of the angiotensin II type 2 receptor AT2R is a novel therapeutic strategy for glioblastoma. Unpublished Work. 2022.
- Beaulieu, J.-M.; Gainetdinov, R.R. The Physiology, Signaling, and Pharmacology of Dopamine Receptors. Pharmacol. Rev. 2011, 63, 182–217.
- Vallone, D.; Picetti, R.; Borrelli, E. Structure and function of dopamine receptors. Neurosci. Biobehav. Rev. 2000, 24, 125–132.
- Borasio, G.D.; Linke, R.; Schwarz, J.; Schlamp, V.; Abel, A.; Mozley, P.D.; Tatsch, K. Dopaminergic deficit in amyotrophic lateral sclerosis assessed with [I-123] IPT single photon emission computed tomography. J. Neurol. Neurosurg. Psychiatry 1998, 65, 263–265.
- Lai, C.-Y.; Liu, Y.-J.; Lai, H.-L.; Chen, H.-M.; Kuo, H.-C.; Liao, Y.-P.; Chern, Y. The D2 Dopamine Receptor Interferes With the Protective Effect of the A2A Adenosine Receptor on TDP-43 Mislocalization in Experimental Models of Motor Neuron Degeneration. Front. Neurosci. 2018, 12, 187.
- Huang, X.; Roet, K.C.; Zhang, L.; Brault, A.; Berg, A.P.; Jefferson, A.B.; Klug-McLeod, J.; Leach, K.L.; Vincent, F.; Yang, H.; et al. Human amyotrophic lateral sclerosis excitability phenotype screen: Target discovery and validation. Cell Rep. 2021, 35, 109224.
- Fujimori, K.; Ishikawa, M.; Otomo, A.; Atsuta, N.; Nakamura, R.; Akiyama, T.; Hadano, S.; Aoki, M.; Saya, H.; Sobue, G.; et al. Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nat. Med. 2018, 24, 1579–1589.
- Huang, X.; Roet, K.C.; Zhang, L.; Brault, A.; Berg, A.P.; Jefferson, A.B.; Klug-McLeod, J.; Leach, K.L.; Vincent, F.; Yang, H.; et al. Human amyotrophic lateral sclerosis excitability phenotype screen: Target discovery and validation. Cell Rep. 2021, 35, 109224.
- Dooley, M.; Markham, A. Pramipexole. Drugs Aging 1998, 12, 495–514.
- Gribkoff, V.K.; Bozik, M.E. KNS-760704 [(6R)-4,5,6,7-tetrahydro-N6-propyl-2,6-benzothiazole-diamine dihydrochloride monohydrate] for the Treatment of Amyotrophic Lateral Sclerosis. CNS Neurosci. Ther. 2008, 14, 215–226.
- Kingwell, K. Dexpramipexole shows promise for ALS in phase II trial. Nat. Rev. Neurol. 2011, 8, 4.
- Cudkowicz, M.E.; Berg, L.H.V.D.; Shefner, J.M.; Mitsumoto, H.; Mora, J.S.; Ludolph, A.; Hardiman, O.; Bozik, M.E.; Ingersoll, E.W.; Archibald, D.; et al. Dexpramipexole versus placebo for patients with amyotrophic lateral sclerosis (EMPOWER): A randomised, double-blind, phase 3 trial. Lancet Neurol. 2013, 12, 1059–1067.
- Zhuang, Y.; Xu, P.; Mao, C.; Wang, L.; Krumm, B.; Zhou, X.E.; Huang, S.; Liu, H.; Cheng, X.; Huang, X.-P.; et al. Structural insights into the human D1 and D2 dopamine receptor signaling complexes. Cell 2021, 184, 931–942.e18.
- Xu, P.; Huang, S.; Mao, C.; Krumm, B.E.; Zhou, X.E.; Tan, Y.; Huang, X.-P.; Liu, Y.; Shen, D.-D.; Jiang, Y.; et al. Structures of the human dopamine D3 receptor-Gi complexes. Mol. Cell 2021, 81, 1147–1159.e4.
- Wang, S.; Wacker, D.; Levit, A.; Che, T.; Betz, R.M.; McCorvy, J.D.; Venkatakrishnan, A.J.; Huang, X.-P.; Dror, R.O.; Shoichet, B.K.; et al. D4 dopamine receptor high-resolution structures enable the discovery of selective agonists. Science 2017, 358, 381–386.
- Nichols, D.E.; Nichols, C.D. Serotonin Receptors. Chem. Rev. 2008, 108, 1614–1641.
- Frazer, A.; Hensler, J.G. Serotonin receptors. In Basic Neurochemistry: Molecular, Cellular and Medical Aspects, 6th ed.; Siegel, G.J., Agranoff, B.W., Albers, R.W., Eds.; Lippincott-Raven: Philadelphia, PA, USA, 1999.
- El Oussini, H.; Bayer, H.; Scekic-Zahirovic, J.; Vercruysse, P.; Sinniger, J.; Dirrig-Grosch, S.; Dieterlé, S.; Echaniz-Laguna, A.; Larmet, Y.; Müller, K.; et al. Serotonin 2B receptor slows disease progression and prevents degeneration of spinal cord mononuclear phagocytes in amyotrophic lateral sclerosis. Acta Neuropathol. 2016, 131, 465–480.
- Dentel, C.; Palamiuc, L.; Henriques, A.; Lannes, B.; Spreux-Varoquaux, O.; Gutknecht, L.; René, F.; Echaniz-Laguna, A.; De Aguilar, J.-L.G.; Lesch, K.P.; et al. Degeneration of serotonergic neurons in amyotrophic lateral sclerosis: A link to spasticity.
- Arnoux, A.; Ayme-Dietrich, E.; Dieterle, S.; Goy, M.-A.; Schann, S.; Frauli, M.; Monassier, L.; Dupuis, L. Evaluation of a 5-HT2B receptor agonist in a murine model of amyotrophic lateral sclerosis. Sci. Rep. 2021, 11, 23582.
- Elangbam, C.S.; Job, L.E.; Zadrozny, L.M.; Barton, J.C.; Yoon, L.W.; Gates, L.D.; Slocum, N. 5-Hydroxytryptamine (5HT)-induced valvulopathy: Compositional valvular alterations are associated with 5HT2B receptor and 5HT transporter transcript changes in Sprague-Dawley rats. Exp. Toxicol. Pathol. 2008, 60, 253–262
- Lacomblez, L.; Bensimon, G.; Douillet, P.; Doppler, V.; Salachas, F.; Meininger, V. Xaliproden in amyotrophic lateral sclerosis: Early clinical trials. Amyotroph. Lateral Scler. 2004, 5, 99–106.
- Xu, P.; Huang, S.; Zhang, H.; Mao, C.; Zhou, X.E.; Cheng, X.; Simon, I.A.; Shen, D.-D.; Yen, H.-Y.; Robinson, C.V.; et al. Structural insights into the lipid and ligand regulation of serotonin receptors. Nature 2021, 592, 469–473.
- McCorvy, J.D.; Wacker, D.; Wang, S.; Agegnehu, B.; Liu, J.; Lansu, K.; Tribo, A.R.; Olsen, R.H.J.; Che, T.; Jin, J.; et al. Structural determinants of 5-HT2B receptor activation and biased agonism. Nat. Struct. Mol. Biol. 2018, 25, 787–796.
- Peng, Y.; McCorvy, J.D.; Harpsøe, K.; Lansu, K.; Yuan, S.; Popov, P.; Qu, L.; Pu, M.; Che, T.; Nikolajsen, L.F.; et al. 5-HT2C Receptor Structures Reveal the Structural Basis of GPCR Polypharmacology. Cell 2018, 172, 719–730.e14.
- Marucci, G.; Ben, D.D.; Lambertucci, C.; Navia, A.M.; Spinaci, A.; Volpini, R.; Buccioni, M. GPR17 receptor modulators and their therapeutic implications: Review of recent patents. Expert Opin. Ther. Patents 2019, 29, 85–95.
- Dziedzic, A.; Miller, E.; Saluk-Bijak, J.; Bijak, M. The GPR17 Receptor—A Promising Goal for Therapy and a Potential Marker of the Neurodegenerative Process in Multiple Sclerosis. Int. J. Mol. Sci. 2020, 21, 1852.
- Marucci, G.; Ben, D.D.; Lambertucci, C.; Santinelli, C.; Spinaci, A.; Thomas, A.; Volpini, R.; Buccioni, M. The G Protein-Coupled Receptor GPR17: Overview and Update. ChemMedChem 2016, 11, 2567–2574.
- Lecca, D.; Raffaele, S.; Abbracchio, M.P.; Fumagalli, M. Regulation and signaling of the GPR17 receptor in oligodendroglial cells. Glia 2020, 68, 1957–1967.
- Zhao, B.; Zhao, C.; Zhang, X.; Huang, X.; Shi, W.; Fang, S.; Lu, Y.; Zhang, W.; Xia, Q.; Wei, E. The new P2Y-like receptor G protein-coupled receptor 17 mediates acute neuronal injury and late microgliosis after focal cerebral ischemia in rats. Neuroscience 2011, 202, 42–57.
- Bonfanti, E.; Bonifacino, T.; Raffaele, S.; Milanese, M.; Morgante, E.; Bonanno, G.; Abbracchio, M.P.; Fumagalli, M. Abnormal Upregulation of GPR17 Receptor Contributes to Oligodendrocyte Dysfunction in SOD1 G93A Mice. Int. J. Mol. Sci. 2020, 21, 2395.
- Merten, N.; Fischer, J.; Simon, K.; Zhang, L.; Schröder, R.; Peters, L.; Letombe, A.-G.; Hennen, S.; Schrage, R.; Bödefeld, T.; et al. Repurposing HAMI3379 to Block GPR17 and Promote Rodent and Human Oligodendrocyte Differentiation. Cell Chem. Biol. 2018, 25, 775–786.e5.
- Raffaele, S.; Boccazzi, M.; Fumagalli, M. Oligodendrocyte Dysfunction in Amyotrophic Lateral Sclerosis: Mechanisms and Therapeutic Perspectives. Cells 2021, 10, 565.
- Jin, S.; Wang, X.; Xiang, X.; Wu, Y.; Hu, J.; Li, Y.; Dong, Y.L.; Tan, Y.; Wu, X. Inhibition of GPR17 with cangrelor improves cognitive impairment and synaptic deficits induced by Aβ1–42 through Nrf2/HO-1 and NF-κB signaling pathway in mice. Int. Immunopharmacol. 2021, 101, 108335.
- Marschallinger, J.; Schäffner, I.; Klein, B.; Gelfert, R.; Rivera, F.J.; Illes, S.; Grassner, L.; Janssen, M.; Rotheneichner, P.; Schmuckermair, C.; et al. Structural and functional rejuvenation of the aged brain by an approved anti-asthmatic drug. Nat. Commun. 2015, 6, 8466.
- Burnstock, G. An introduction to the roles of purinergic signalling in neurodegeneration, neuroprotection and neuroregeneration. Neuropharmacology 2016, 104, 4–17.
- Insel, P.A. Adrenergic Receptors—Evolving Concepts and Clinical Implications. N. Engl. J. Med. 1996, 334, 580–585.
- Subbarao, K.V.; Hertz, L. Effect of adrenergic agonists on glycogenolysis in primary cultures of astrocytes. Brain Res. 1990, 536, 220–226.
- Patel, M.; Shaw, D. A review of standard pharmacological therapy for adult asthma—Steps 1 to 5. Chronic Respir. Dis. 2015, 12, 165–176.
- Bartus, R.T.; Bétourné, A.; Basile, A.; Peterson, B.L.; Glass, J.; Boulis, N.M. β 2 -Adrenoceptor agonists as novel, safe and potentially effective therapies for Amyotrophic lateral sclerosis (ALS). Neurobiol. Dis. 2016, 85, 11–24.
- Teng, Y.D.; Choi, H.; Huang, W.; Onario, R.C.; Frontera, W.R.; Snyder, E.Y.; Sabharwal, S. Therapeutic effects of clenbuterol in a murine model of amyotrophic lateral sclerosis. Neurosci. Lett. 2006, 397, 155–158.
- Yang, F.; Ling, S.; Zhou, Y.; Zhang, Y.; Lv, P.; Liu, S.; Fang, W.; Sun, W.; A Hu, L.; Zhang, L.; et al. Different conformational responses of the β2-adrenergic receptor-Gs complex upon binding of the partial agonist salbutamol or the full agonist isoprenaline. Natl. Sci. Rev. 2020, 8.
- Parsons, M.E.; Ganellin, C.R. Histamine and its receptors. Br. J. Pharmacol. 2006, 147 (Suppl. 1), S127–S135
- Tiligada, E.; Ennis, M. Histamine pharmacology: From Sir Henry Dale to the 21st century. J. Cereb. Blood Flow Metab. 2018, 177, 469–489.
- Apolloni, S.; Fabbrizio, P.; Amadio, S.; Napoli, G.; Verdile, V.; Morello, G.; Iemmolo, R.; Aronica, E.; Cavallaro, S.; Volonté, C. Histamine Regulates the Inflammatory Profile of SOD1-G93A Microglia and the Histaminergic System Is Dysregulated in Amyotrophic Lateral Sclerosis. Front. Immunol. 2017, 8, 1689.
- Volonté, C.; Apolloni, S.; Sabatelli, M. Histamine beyond its effects on allergy: Potential therapeutic benefits for the treatment of Amyotrophic Lateral Sclerosis (ALS). Pharmacol. Ther. 2019, 202, 120–131.
- Zhang, W.; Zhang, X.; Zhang, Y.; Qu, C.; Zhou, X.; Zhang, S. Histamine Induces Microglia Activation and the Release of Proinflammatory Mediators in Rat Brain Via H1R or H4R. J. Neuroimmune Pharmacol. 2019, 15, 280–291.
- Apolloni, S.; Fabbrizio, P.; Parisi, C.; Amadio, S.; Volonté, C. Clemastine Confers Neuroprotection and Induces an Anti-Inflammatory Phenotype in SOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2014, 53, 518–531.
- Xia, R.; Wang, N.; Xu, Z.; Lu, Y.; Song, J.; Zhang, A.; Guo, C.; He, Y. Cryo-EM structure of the human histamine H1 receptor/Gq complex. Nat. Commun. 2021, 12, 1–9.
- DeepMind. The AlphaFold Database. Available online: https://alphafold.ebi.ac.uk/ (accessed on 22 March 2022).
- Zou, S.; Kumar, U. Cannabinoid Receptors and the Endocannabinoid System: Signaling and Function in the Central Nervous System. Int. J. Mol. Sci. 2018, 19, 833.
- Kaminski, N.E. Immune regulation by cannabinoid compounds through the inhibition of the cyclic AMP signaling cascade and altered gene expression. Biochem. Pharmacol. 1996, 52, 1133–1140.
- Mazzon, E.; Giacoppo, S. Can cannabinoids be a potential therapeutic tool in amyotrophic lateral sclerosis? Neural Regen. Res. 2016, 11, 1896–1899.
- Urbi, B.; Owusu, M.A.; Hughes, I.; Katz, M.; Broadley, S.; Sabet, A. Effects of cannabinoids in Amyotrophic Lateral Sclerosis (ALS) murine models: A systematic review and meta-analysis. J. Neurochem. 2018, 149, 284–297.
- Shoemaker, J.L.; Seely, K.A.; Reed, R.L.; Crow, J.P.; Prather, P.L. The CB2 cannabinoid agonist AM-1241 prolongs survival in a transgenic mouse model of amyotrophic lateral sclerosis when initiated at symptom onset. J. Neurochem. 2006, 101, 87–98.
- Kim, K.; Moore, D.H.; Makriyannis, A.; Abood, M.E. AM1241, a cannabinoid CB2 receptor selective compound, delays disease progression in a mouse model of amyotrophic lateral sclerosis. Eur. J. Pharmacol. 2006, 542, 100–105.
- Bilsland, L.G.; Dick, J.R.T.; Pryce, G.; Petrosino, S.; Di Marzo, V.; Baker, D.; Greensmith, L. Increasing cannabinoid levels by pharmacological and genetic manipulation delays disease progression in SOD1 mice. FASEB J. 2006, 20, 1003–1005.
- Hua, T.; Li, X.; Wu, L.; Iliopoulos-Tsoutsouvas, C.; Wang, Y.; Wu, M.; Shen, L.; Brust, C.A.; Nikas, S.P.; Song, F.; et al. Activation and Signaling Mechanism Revealed by Cannabinoid Receptor-Gi Complex Structures. Cell 2020, 180, 655–665.e18.
- Xing, C.; Zhuang, Y.; Xu, T.-H.; Feng, Z.; Zhou, X.E.; Chen, M.; Wang, L.; Meng, X.; Xue, Y.; Wang, J.; et al. Cryo-EM Structure of the Human Cannabinoid Receptor CB2-Gi Signaling Complex. Cell 2020, 180, 645–654.e13.
- Reader, J.; Holt, D.; Fulton, A. Prostaglandin E2 EP receptors as therapeutic targets in breast cancer. Cancer Metastasis Rev. 2011, 30, 449–463.
- Johansson, T.; Narumiya, S.; Zeilhofer, H.U. Contribution of peripheral versus central EP1 prostaglandin receptors to inflammatory pain. Neurosci. Lett. 2011, 495, 98–101.
- Fedyk, E.R.; Phipps, R.P. Prostaglandin E2 receptors of the EP2 and EP4 subtypes regulate activation and differentiation of mouse B lymphocytes to IgE-secreting cells. Proc. Natl. Acad. Sci. USA 1996, 93, 10978–10983.
- Watanabe, K.; Kawamori, T.; Nakatsugi, S.; Ohta, T.; Ohuchida, S.; Yamamoto, H.; Maruyama, T.; Kondo, K.; Ushikubi, F.; Narumiya, S.; et al. Role of the prostaglandin E receptor subtype EP1 in colon carcinogenesis. Cancer Res. 1999, 59, 5093–5096.
- Sun, X.; Li, Q. Prostaglandin EP2 receptor: Novel therapeutic target for human cancers (Review). Int. J. Mol. Med. 2018, 42, 1203–1214.
- Takeuchi, K.; Kato, S.; Amagase, K. Prostaglandin EP Receptors Involved in Modulating Gastrointestinal Mucosal Integrity. J. Pharmacol. Sci. 2010, 114, 248–261.
- Yang, T.; Du, Y. Distinct Roles of Central and Peripheral Prostaglandin E2 and EP Subtypes in Blood Pressure Regulation. Am. J. Hypertens. 2012, 25, 1042–1049.
- Friedman, E.A.; Ogletree, M.L.; Haddad, E.V.; Boutaud, O. Understanding the role of prostaglandin E2 in regulating human platelet activity in health and disease. Thromb. Res. 2015, 136, 493–503.
- Takasaki, I.; Nojima, H.; Shiraki, K.; Sugimoto, Y.; Ichikawa, A.; Ushikubi, F.; Narumiya, S.; Kuraishi, Y. Involvement of cyclooxygenase-2 and EP3 prostaglandin receptor in acute herpetic but not postherpetic pain in mice. Neuropharmacology 2005, 49, 283–292.
- Xu, S.; Zhou, W.; Ge, J.; Zhang, Z. Prostaglandin E2 receptor EP4 is involved in the cell growth and invasion of prostate cancer via the cAMP-PKA/PI3K-Akt signaling pathway. Mol. Med. Rep. 2018, 17, 4702–4712.
- Iłżecka, J. Prostaglandin E2 is increased in amyotrophic lateral sclerosis patients. Acta Neurol. Scand. 2003, 108, 125–129.
- Kosuge, Y.; Nango, H.; Kasai, H.; Yanagi, T.; Mawatari, T.; Nishiyama, K.; Miyagishi, H.; Ishige, K.; Ito, Y. Generation of Cellular Reactive Oxygen Species by Activation of the EP2 Receptor Contributes to Prostaglandin E2-Induced Cytotoxicity in Motor Neuron-Like NSC-34 Cells. Oxidative Med. Cell. Longev. 2020, 2020, 1–14.
- Bilak, M.; Wu, L.; Wang, Q.; Haughey, N.; Conant, K.; St. Hillaire, C.; Andreasson, K. PGE2 receptors rescue motor neurons in a model of amyotrophic lateral sclerosis. Ann. Neurol. 2004, 56, 240–248.
- Qu, C.; Mao, C.; Xiao, P.; Shen, Q.; Zhong, Y.-N.; Yang, F.; Shen, D.-D.; Tao, X.; Zhang, H.; Yan, X.; et al. Ligand recognition, unconventional activation, and G protein coupling of the prostaglandin E2 receptor EP2 subtype. Sci. Adv. 2021, 7, eabf1268.
- Morimoto, K.; Suno, R.; Hotta, Y.; Yamashita, K.; Hirata, K.; Yamamoto, M.; Narumiya, S.; Iwata, S.; Kobayashi, T. Crystal structure of the endogenous agonist-bound prostanoid receptor EP3. Nat. Chem. Biol. 2018, 15, 8–10.
- Nojima, S.; Fujita, Y.; Kimura, K.T.; Nomura, N.; Suno, R.; Morimoto, K.; Yamamoto, M.; Noda, T.; Iwata, S.; Shigematsu, H.; et al. Cryo-EM Structure of the Prostaglandin E Receptor EP4 Coupled to G Protein. Structure 2020, 29, 252–260.e6.
- Umetsu, Y.; Tenno, T.; Goda, N.; Shirakawa, M.; Ikegami, T.; Hiroaki, H. Structural difference of vasoactive intestinal peptide in two distinct membrane-mimicking environments. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2011, 1814, 724–730.
- Harmar, A.J.; Fahrenkrug, J.; Gozes, I.; Laburthe, M.; May, V.; Pisegna, J.R.; Vaudry, D.; Vaudry, H.; A Waschek, J.; I Said, S. Pharmacology and functions of receptors for vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide: IUPHAR Review 1. J. Cereb. Blood Flow Metab. 2012, 166, 4–17
- Couvineau, A.; Laburthe, M. VPAC receptors: Structure, molecular pharmacology and interaction with accessory proteins. J. Cereb. Blood Flow Metab. 2012, 166, 42–50.
- Duan, J.; Shen, D.-D.; Zhou, X.E.; Bi, P.; Liu, Q.-F.; Tan, Y.-X.; Zhuang, Y.-W.; Zhang, H.-B.; Xu, P.-Y.; Huang, S.-J.; et al. Cryo-EM structure of an activated VIP1 receptor-G protein complex revealed by a NanoBiT tethering strategy. Nat. Commun. 2020, 11, 1–10.
- Harmar, A.J.; Fahrenkrug, J.; Gozes, I.; Laburthe, M.; May, V.; Pisegna, J.R.; Vaudry, D.; Vaudry, H.; A Waschek, J.; I Said, S. Pharmacology and functions of receptors for vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide: IUPHAR Review 1. J. Cereb. Blood Flow Metab. 2012, 166, 4–17.
- Staines, D.R. Are multiple sclerosis and amyotrophic lateral sclerosis autoimmune disorders of endogenous vasoactive neuropeptides? Med. Hypotheses 2008, 70, 413–418.
- Iwasaki, Y.; Ikeda, K.; Ichikawa, Y.; Igarashi, O. Vasoactive intestinal peptide influences neurite outgrowth in cultured rat spinal cord neurons. Neurol. Res. 2001, 23, 851–854.
- Solés-Tarrés, I.; Cabezas-Llobet, N.; Vaudry, D.; Xifró, X. Protective Effects of Pituitary Adenylate Cyclase-Activating Polypeptide and Vasoactive Intestinal Peptide against Cognitive Decline in Neurodegenerative Diseases. Front. Cell. Neurosci. 2020, 14, 221.
- Waschek, J. VIP and PACAP: Neuropeptide modulators of CNS inflammation, injury, and repair. J. Cereb. Blood Flow Metab. 2013, 169, 512–523.
- Martínez, C.; Juarranz, Y.; Gutiérrez-Cañas, I.; Carrión, M.; Pérez-García, S.; Villanueva-Romero, R.; Castro, D.; Lamana, A.; Mellado, M.; González-Álvaro, I.; et al. A Clinical Approach for the Use of VIP Axis in Inflammatory and Autoimmune Diseases. Int. J. Mol. Sci. 2019, 21, 65.
- Niswender, C.M.; Conn, P.J. Metabotropic Glutamate Receptors: Physiology, Pharmacology, and Disease. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 295–322.
- Anneser, J.M.H.; Chahli, C.; Ince, P.G.; Borasio, G.D.; Shaw, P. Glial Proliferation and Metabotropic Glutamate Receptor Expression in Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol. 2004, 63, 831–840.
- Milanese, M.; Giribaldi, F.; Melone, M.; Bonifacino, T.; Musante, I.; Carminati, E.; Rossi, P.I.; Vergani, L.; Voci, A.; Conti, F.; et al. Knocking down metabotropic glutamate receptor 1 improves survival and disease progression in the SOD1G93A mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2014, 64, 48–59.
- Rossi, D.; Brambilla, L.; Valori, C.F.; Roncoroni, C.; Crugnola, A.; Yokota, T.; Bredesen, D.E.; Volterra, A. Focal degeneration of astrocytes in amyotrophic lateral sclerosis. Cell Death Differ. 2008, 15, 1691–1700.
- Anneser, J.; Chahli, C.; Borasio, G. Protective effect of metabotropic glutamate receptor inhibition on amyotrophic lateral sclerosis–cerebrospinal fluid toxicity in vitro. Neuroscience 2006, 141, 1879–1886.
- Crupi, R.; Impellizzeri, D.; Cuzzocrea, S. Role of Metabotropic Glutamate Receptors in Neurological Disorders. Front. Mol. Neurosci. 2019, 12, 20.
- Zhang, J.; Qu, L.; Wu, L.; Tang, X.; Luo, F.; Xu, W.; Xu, Y.; Liu, Z.-J.; Hua, T. Structural insights into the activation initiation of full-length mGlu1. Protein Cell 2020, 12, 662–667.
- Seven, A.B.; Barros-Álvarez, X.; de Lapeyrière, M.; Papasergi-Scott, M.M.; Robertson, M.J.; Zhang, C.; Nwokonko, R.M.; Gao, Y.; Meyerowitz, J.G.; Rocher, J.-P.; et al. G-protein activation by a metabotropic glutamate receptor. Nature 2021, 595, 450–454.
- Lin, S.; Han, S.; Cai, X.; Tan, Q.; Zhou, K.; Wang, D.; Wang, X.; Du, J.; Yi, C.; Chu, X.; et al. Structures of Gi-bound metabotropic glutamate receptors mGlu2 and mGlu4. Nature 2021, 594, 583–588.
More
Information
Subjects:
Medicine, Research & Experimental
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
978
Revisions:
6 times
(View History)
Update Date:
12 May 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No