Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Niribili Sarmah | -- | 2146 | 2022-05-06 21:57:58 | | | |
| 2 | Rita Xu | Meta information modification | 2146 | 2022-05-07 04:15:48 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Sarmah, N.; Nauli, S.; Nauli, A.; Ally, A. Nitric Oxide Synthase. Encyclopedia. Available online: https://encyclopedia.pub/entry/22674 (accessed on 23 July 2026).
Sarmah N, Nauli S, Nauli A, Ally A. Nitric Oxide Synthase. Encyclopedia. Available at: https://encyclopedia.pub/entry/22674. Accessed July 23, 2026.
Sarmah, Niribili, Surya Nauli, Andromeda Nauli, Ahmmed Ally. "Nitric Oxide Synthase" Encyclopedia, https://encyclopedia.pub/entry/22674 (accessed July 23, 2026).
Sarmah, N., Nauli, S., Nauli, A., & Ally, A. (2022, May 06). Nitric Oxide Synthase. In Encyclopedia. https://encyclopedia.pub/entry/22674
Sarmah, Niribili, et al. "Nitric Oxide Synthase." Encyclopedia. Web. 06 May, 2022.
Copy Citation
Nitric oxide synthase (NOS) plays important roles within the cardiovascular system in physiological states as well as in pathophysiologic and specific cardiovascular (CV) disease states, such as hypertension (HTN), arteriosclerosis, and cerebrovascular accidents.
nociception
periaqueductal gray matter
hypertension
nitric oxide
nitric oxide synthase
angiotensin-converting enzyme
inflammation
1. Introduction
Endothelial nitric oxide synthase (eNOS) is one of the isoforms of nitric oxide synthase (NOS). Endothelial NOS is highly expressed in vascular endothelial cells. The biochemical reaction to produce nitric oxide is shown below:
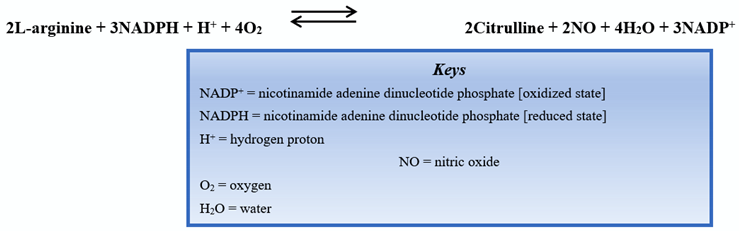
Discovered in 1991 [1], the eNOS enzyme was found to be responsible for the production of nitric oxide (NO), an essential compound in vasodilation responses and a critical regulator of cardiovascular (CV) homeostasis [2]. In addition to its action in vascular endothelial cells, eNOS is also active in hippocampal neurons as well as postganglionic sympathetic and dorsal root ganglion [3]. Nitric oxide increases the activity of guanylyl cyclase, an enzyme responsible for producing cyclic guanosine monophosphate (cGMP) that promotes smooth muscle relaxation. Endothelial NOS maintains a healthy CV system (CVS), partly by regulating and maintaining vascular tone: migration, production, and maturation of cells; leukocyte adhesion; and platelet aggregation [4]. A schematic of NO production by the endothelial cells of the vasculature is shown in Figure 1.
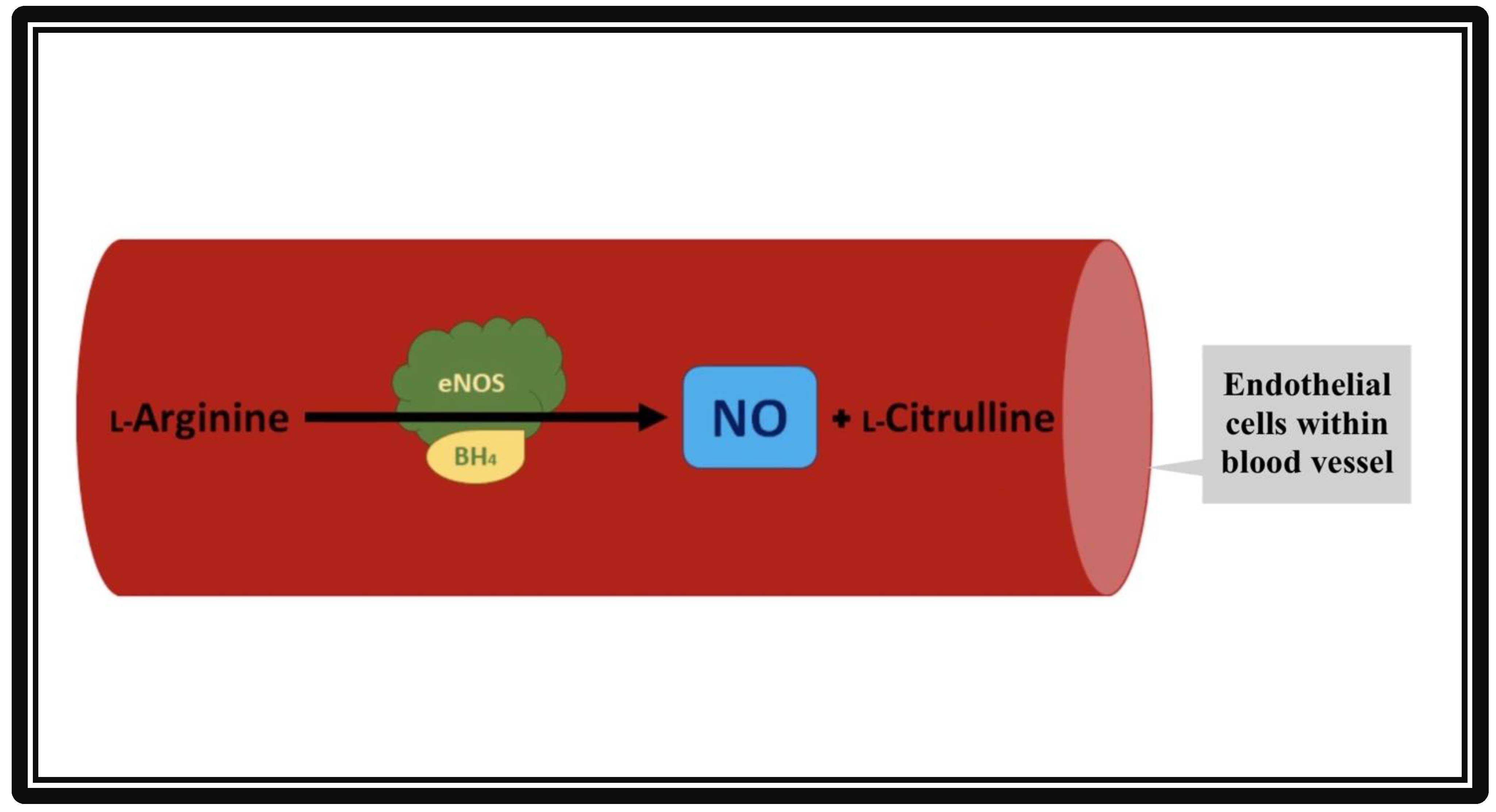
Figure 1. Formation of nitric oxide (NO) from L-Arginine in the presence of co-factor tetrahydropbiopterin (BH4) and endothelial nitric oxide synthase (eNOS). The vascular endothelial cells produce NO by utilizing eNOS. The NO then diffuses from vascular endothelia to vascular smooth muscle cells. Inside the smooth muscles, NO activates guanylate cyclase, which converts GTP to cGMP. cGMP relaxes the vascular smooth muscle cells, resulting in vasodilation.
1.1. Nociception
This manuscript’s primary focus is to elaborate the roles of eNOS in relationship to nociception and CV system. Nociception is described as a subjective sensory and emotional experience that is accompanied by some degree of tissue damage. Stimuli of a painful nature may result from different inputs including mechanical, chemical, or thermal origins. Mechanical responses include injury such as a laceration to the skin [5], while chemical stimuli may include events such as exposure to a “hot” gustatory stimulus, such as capsaicin, on the tongue [6]. In addition, thermal stimuli include contact with extreme heat or extreme cold [7]. These phenomena may stimulate sensory pain receptors that will relay signals to different areas of the brain via afferent fibers in the peripheral and central nervous system. These signals are integrated by sensory integration centers involving the medulla oblongata, periaqueductal gray matter (PAG), hypothalamus, and thalamus [8]. The aforementioned relay pathways are relevant to the physiological role played by eNOS. Stimulation of nociceptors may activate the eNOS pathway via autonomic nervous system-mediated inflammation and vasodilation cascades [9]. The CVS plays a significant role in the appropriate execution of such cascades. Accordingly, researchers will discuss these pathways in depth, including the function of eNOS in relation to CV pathologies, such as hypertension and atherosclerosis.
1.2. Cardiovascular System
The CV system is a complex arrangement of arteries, capillaries, veins, and a central organ or the heart that works systematically to circulate blood throughout the body. It is a critical system that drives the transportation of vital resources and signals to different areas in the body, including oxygen, carbon dioxide, vitamins, minerals, neurotransmitters (for platelet aggregation), insulin, and NO [10]. The eNOS isoenzyme can interact with the cardiovascular system by regulating signal relay and resource allocation. However, the exact mechanism delineating this relationship with nociception remains unknown. For example, reduced function or bioavailability of the eNOS isoenzyme is linked to increased risk of essential hypertension, preeclampsia, diabetic nephropathy, retinopathy, migraine, and erectile dysfunction in humans [11]. As noted, reduced eNOS expression produces several deleterious effects; however, eNOS upregulation can result in homeostatic dysregulation as well [12]. Overexpression in the rostral ventrolateral medulla (RVLM) is positively correlated with hypotension and bradycardia. Both pathologies are preceded by altered neurotransmitter levels, namely increased levels of inhibitory gamma amino-butyric acid (GABA) and decreased levels of excitatory glutamate [12]. Upregulation of eNOS may be a result of stress caused by the release of acetylcholine (ACh), bradykinin, histamine, and 17β-estradiol-mediated phosphorylation [13].
2. Endothelial Nitric Oxide Synthase and Nociception
Nociception, or more commonly known as pain, is a sensation that also involves an emotional component. The International Association for the Study of Pain (IASP9) offers the following definition of pain: “An unpleasant sensory and emotional experience associated with actual or potential tissue damage or described in terms of such damage” [14]. Researchers have previously published an extensive review regarding the role of nNOS isoform on CV responses elicited during mechanically, heat-, and cold-induced changes in mean arterial pressure (MAP) and heart rate (HR) using a model of anesthetized rat [9]. Briefly, the alterations in CV responses due to pain are dependent on the type of pain, intensity of pain, involvement of specific nociceptors, neural pathways, and the involvement of various neurotransmitters or receptors [15]. It has been widely studied that the eNOS system interacts with the ventrolateral medullar (VLM) and PAG to modulate pain. Moreover, several brain regions such as the RVLM, the caudal ventrolateral medulla (CVLM), and the dorsolateral periaqueductal gray matter (dlPAG) play differential roles in integrating and modulating CV responses during pain [13]. Manipulating NO concentrations within the dlPAG or medulla oblongata affects the glutamatergic and GABAergic pathways during nociceptive experiences triggered by thermal (heat/cold) stimuli but not mechanical stimuli or pinch/pressure [16]. Past studies demonstrate that dysregulation of all NOS isoforms (nNOS, eNOS, inducible NOS (iNOS)) can significantly alter the experience of pain. For example, overproduction of NO by all NOS isoforms may result in neuroprotective or neurotoxic impacts mediated through the generation of reactive oxygen waste products (peroxynitrite) [17]. Subsequent chain reactions with carbon dioxide (CO2) can induce the destruction of neurons by way of oxidizing lipids, denaturing polypeptides, and destabilizing structural bases in DNA [17].
While substantial evidence regarding the involvement of nNOS and iNOS in producing neuropathic pain has been uncovered, the relationship between eNOS and nociception remains unexplored due to their complicated and irregular interactions [9]. Undoubtedly, NO is implicated in both pro-nociception as well as anti-nociception [18]. Several studies find inconsistent results regarding the effect of arginine bioavailability on the pain status of the participants. One such study evaluates this relationship in both chronic and acute pain conditions related to sickle cell disease (SCD) [19]. The reduced NO from SCD-related hemolysis and oxidative stress is noted to result in endothelial dysfunction during experiences of vaso-occlusive pain [19]. This dynamic is consistent throughout varying participant demographics, including adults and children. Another study appraises the eNOS-nociceptive pathway in rats with chronic post-ischemia pain (CPIP) through norepinephrine-induced nociception and vasoconstrictor hypersensitivity [20]. Chronic post-ischemia pain (CPIP+) rats and control rats received intradermal injection of either vasopressin or the eNOS inhibitor (N5)-(1-Iminoethyl)-L-ornithine dihydrochloride (L-NIO), and their nociceptive responses were compared [20]. Nociceptive responses were produced at significant levels (p < 0.01) in rats injected with L-NIO, suggesting that eNOS-facilitated vascular dilatation may attenuate pain responses [20]. Please see Figure 2 for the proposed mechanism.
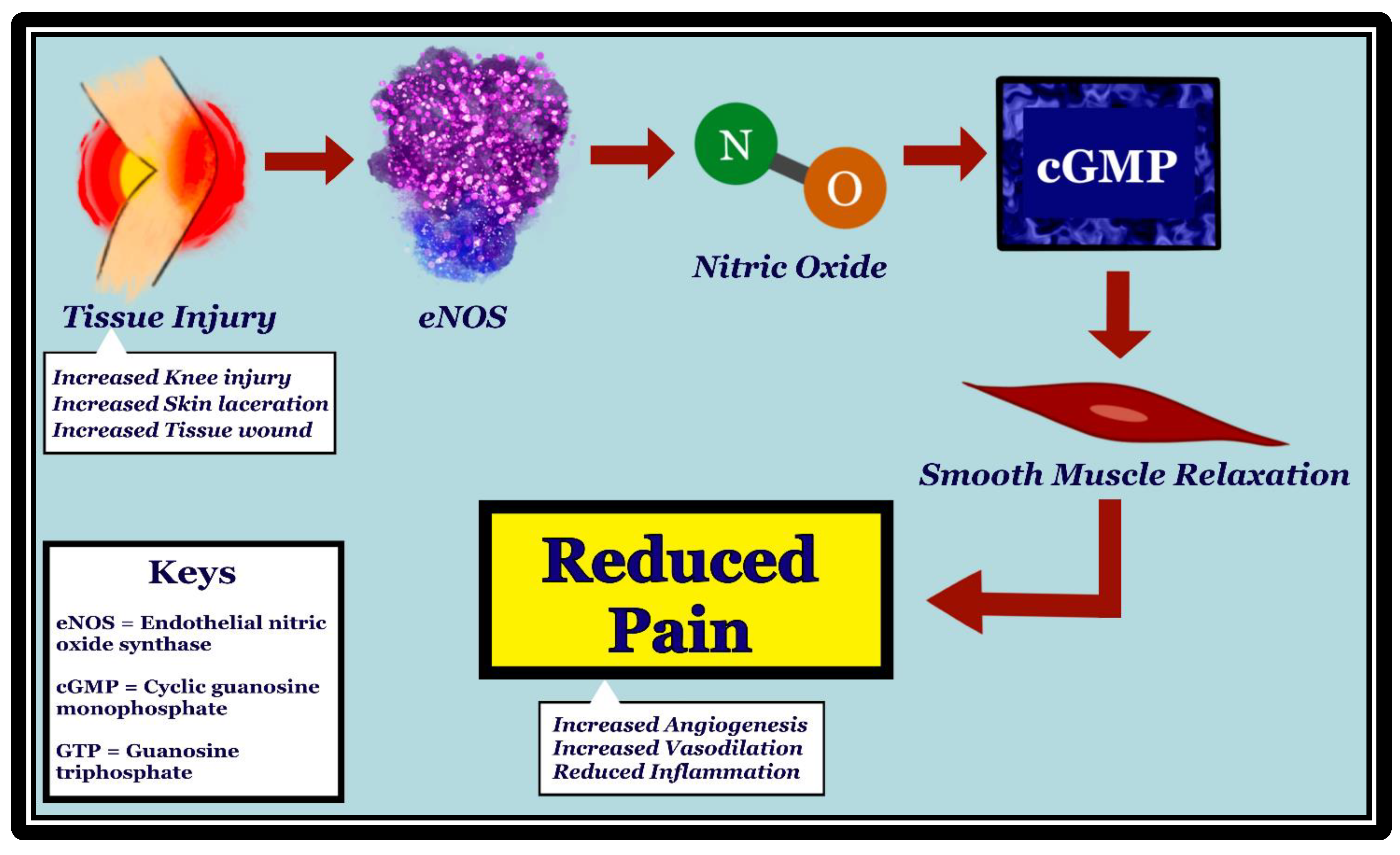
Figure 2. Mechanism of how initial tissue injury leads to pain reduction through activation of eNOS, production of nitric oxide (NO), and increase in cGMP. eNOS is stimulated in vascular endothelia by tissue injury, such as knee injury, skin laceration, and wounds. NO, which is produced by eNOS, diffuses to the vascular smooth muscle cells. Upon stimulation by NO, guanylate cyclase converts GTP to cGMP, resulting in vascular smooth muscle relaxation. Vasodilation has been shown to be associated with pain reduction, possibly through reduced inflammation and increased angiogenesis.
Using a chronic constriction injury (CCI) rat model, Chu et al. showed that atorvastatin, a cholesterol-lowering drug, could reduce nociceptive sensitization and thermal hyperalgesia of peripheral neuropathy. It improved the inflammatory activity through reductions in cyclooxygenase-2 (COX-2) and iNOS. In addition, atorvastatin also increased neuroprotective factors that elicit positive responses in angiogenesis, including eNOS, vascular endothelial growth factor (VEGF), and (phosphorylated) protein kinase B (pAkt/Akt). However, this angiogenic property was not observed in eNOS knock-out mice, suggesting the importance of the eNOS-associated signal cascade in promoting vascular growth during nociceptive events [21]. This relationship is in alignment with what researchers know thus far of the VEGF signaling pathway, which has been shown to upregulate expression of Akt and eNOS [22]. Thus, improving vascular permeability can, in turn, aid in the recovery process of injured nervous tissue [23]. The protective role played by eNOS is further supported by its neuroprotective capabilities under conditions of ischemia or shear stress, positing the role of eNOS in earlier states of inflammatory nociception versus a neurotrophic role in later states [3][24]. Specifically, eNOS serves as a messenger to peripheral and central afferent neuronal sensitization following noxious stimuli [25]. Moreover, cluster of differentiation 31 (CD31) immunopositivity assay is shown to increase staining in complete Freund’s adjuvant (CFA) subject vessels, a comparable effect to eNOS that might indicate an interrelation since CD31 can modulate eNOS activity during shear stress via association or dissociation [3]. Evidence in favor of this finding is endorsed by an analysis of the role of chronic prostatitis/pelvic pain syndrome (CP/CPPS) and its relationship with endothelial dysfunction, specifically the expression of eNOS and cGMP levels in experimental autoimmune prostatitis (EAP), a rat model of CP/CPPS [26]. CP/CPPS is an inflammatory condition involving persistent pain and discomfort in the pelvic and genital areas.
3. Endothelial Nitric Oxide Synthase and the Cardiovascular System
In the CV system, eNOS mediates vasodilation via the production of NO. In response to a surplus of L-arginine, the severity of acute episodic pain related to sickle cell anemia (SCD), a chronic anemia exacerbated by CV complications, was reduced [19]. Of note, SCD patients have arginine deficiency. The cell-free hemoglobin from the lysed sickled erythrocytes consume nearby NO via the reaction shown below [27]:
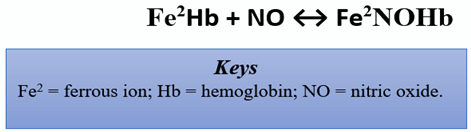
This reaction is an eradicant pathway for NO bioactivity. It proceeds at a rapid rate of 6–8 × 107 per molar per second, rendering NO into the dead-end product, nitrate [28]. Thus, reduced NO is conceivably tied to vaso-occlusive pain and nociceptive processing. Herein, the reduced bioavailability of arginine in such patients is linked with experiences of acute pain, vaso-occlusive pain, endothelial dysfunction, pulmonary complications, risk of leg ulcers, and early mortality [29]. However, the specific mechanism remains unclear, as other studies corroborate increased pain in association with elevated levels of arginine [30].
Individuals carrying the thymine-cytosine and cytosine-cytosine (TC/CC) genotypes and the C allele for the g.-786T > C polymorphisms (eNOS) were more likely to respond better to enalapril, an angiotensin-converting enzyme (ACE) inhibitor. The polymorphism may be responsible for a more robust NO production, conferring the individuals a better response to the antihypertensive drug [31]. Peroxisome proliferator-activated receptor gamma coactivator-1alpha (PGC-1α) has also been shown to enhance NO production and reduce blood pressure. However, it increased NO production not by altering eNOS expression, but by inhibiting the uncoupling of eNOS dimers [32].
Several studies aim to explore the roles of transient receptor potential vanilloid type 1 (TRPV1) on NOS activation and its subsequent downstream effects of CV-specific angiogenesis in primary nociceptive sensory neurons. TRPV1 is mainly distributed throughout vascular endothelial cells, smooth muscle cells, and perivascular nerve cells in the cardiovascular system. It is responsive to inflammatory and noxious stimuli and can serve as a blood pressure mechanoreceptor within vasculature [33]. In addition, NO-dependent processes as a result of TRPV1 stimulation are a vital facet in the creation of new blood vessels and wound-healing, two factors that are important in countering the effects of cardiovascular disease such as arteriosclerosis or ischemic heart disease [34]. Activation of TRPV1 in endothelial cells promotes Ca2+-dependent signaling of phosphatidylinositol-3-kinase (PI3K), Akt, and Ca2+/calmodulin-dependent protein kinase II (CaMKII), which subsequently leads to increases in TRPV1-eNOS complex formation, eNOS activation, and NO production. eNOS and TRPV1 knockouts displayed reduced angiogenesis, increased atherosclerotic lesions, and reduced phosphorylation of eNOS and Akt [35].
Previously, it has been shown that the reduced atherosclerosis through uncoupling activity results in reduction of NO [13]. Studies explore oxidized low-density lipoprotein (Ox-LDL) as a cytotoxic agent that can impair endothelial cells and exacerbate the progression of atherosclerosis. This mechanism is an advancement of oxLDL-mediated inflammation that consequently decreases peroxisome proliferator-activated receptor gamma (PPARγ) endothelium protection, a factor that typically participates in eNOS expression through AMP-activated protein kinase (AMPK) and lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) signal cascades [36]. Inactivation of this pathway can cause detrimental fluctuation of atherosclerotic plaques and vascular malfunction.
Sildenafil (Viagra®, Revatio®) is a phosphodiesterase type 5 inhibitor (PDE5 inhibitor) that induces vasodilation by preventing PDE5 from degrading cGMP. Sildenafil exerts its neuroprotective effect by inhibiting inflammation and demyelination in the cerebellum [37]. Sildenafil was capable of decreasing the expression of the pro-inflammatory cytokines interleukin-1-beta (IL-1β) and tumor necrosis factor alpha (TNF-α); decreasing glial fibrillation acidic protein (GFAP), nuclear factor kappa light chain enhancer of active B-cells (NFkB), inactive AMPK, iNOS; and increasing the anti-inflammatory cytokine IL-10 and nuclear factor alpha of kappa light polypeptide gene enhancer in B-cell inhibitor (IKβα) [37]. Based on this discovery, sildenafil likely induces its anti-inflammatory and neuroprotective effects through the modulation of AMPK–IKβα–NFκB signaling [37].
References
- Pollock, J.S.; Forstermann, U.; Mitchell, J.A.; Warner, T.D.; Schmidt, H.H.; Nakane, M.; Murad, F. Purification and characterization of particulate endothelium-derived relaxing factor synthase from cultured and native bovine aortic endothelial cells. Proc. Natl. Acad. Sci. USA 1991, 88, 10480–10484.
- Ma, T.; Zhang, Z.; Chen, Y.; Su, H.; Deng, X.; Liu, X.; Fan, Y. Delivery of Nitric Oxide in the Cardiovascular System: Implications for Clinical Diagnosis and Therapy. Int. J. Mol. Sci. 2021, 22, 12166.
- Borsani, E.; Giovannozzi, S.; Cocchi, M.A.; Boninsegna, R.; Rezzani, R.; Rodella, L.F. Endothelial nitric oxide synthase in dorsal root ganglia during chronic inflammatory nociception. Cells Tissues Organs 2013, 197, 159–168.
- Forstermann, U.; Munzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714.
- Dreno, B.; Bettoli, V.; Perez, M.; Bouloc, A.; Ochsendorf, F. Cutaneous lesions caused by mechanical injury. Eur. J. Dermatol. 2015, 25, 114–121.
- Frias, B.; Merighi, A. Capsaicin, Nociception and Pain. Molecules 2016, 21, 797.
- Chen, J.; Kandle, P.F.; Murray, I.; Fitzgerald, L.A.; Sehdev, J.S. Physiology, Pain; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK539789/ (accessed on 25 April 2022).
- Mills, E.P.; Keay, K.A.; Henderson, L.A. Brainstem Pain-Modulation Circuitry and Its Plasticity in Neuropathic Pain: Insights From Human Brain Imaging Investigations. Front Pain Res. (Lausanne) 2021, 2, 705345.
- Ally, A.; Powell, I.; Ally, M.M.; Chaitoff, K.; Nauli, S.M. Role of neuronal nitric oxide synthase on cardiovascular functions in physiological and pathophysiological states. Nitric Oxide 2020, 102, 52–73.
- Jain, V.; Bordes, S.; Bhardwaj, A. Physiology, Pulmonary Circulatory System. In StatPearls ; Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK525948/ (accessed on 26 February 2022).
- Albrecht, E.W.; Stegeman, C.A.; Heeringa, P.; Henning, R.H.; van Goor, H. Protective role of endothelial nitric oxide synthase. J. Pathol. 2003, 199, 8–17.
- Ozaki, M.; Kawashima, S.; Yamashita, T.; Hirase, T.; Namiki, M.; Inoue, N.; Hirata, K.; Yasui, H.; Sakurai, H.; Yoshida, Y.; et al. Overexpression of endothelial nitric oxide synthase accelerates atherosclerotic lesion formation in apoE-deficient mice. J. Clin. Investig. 2002, 110, 331–340.
- Tran, N.; Garcia, T.; Aniqa, M.; Ali, S.; Ally, A.; Nauli, S.M. Endothelial Nitric Oxide Synthase (eNOS) and the Cardiovascular System: In Physiology and in Disease States. Am. J. Biomed. Sci. Res. 2022, 15, 153–177.
- Raja, S.N.; Carr, D.B.; Cohen, M.; Finnerup, N.B.; Flor, H.; Gibson, S.; Keefe, F.J.; Mogil, J.S.; Ringkamp, M.; Sluka, K.A.; et al. The revised International Association for the Study of Pain definition of pain: Concepts, challenges, and compromises. Pain 2020, 161, 1976–1982.
- Yam, M.F.; Loh, Y.C.; Tan, C.S.; Khadijah Adam, S.; Abdul Manan, N.; Basir, R. General Pathways of Pain Sensation and the Major Neurotransmitters Involved in Pain Regulation. Int. J. Mol. Sci. 2018, 19, 2164.
- Karlsson, G.A.; Chaitoff, K.A.; Hossain, S.; Bohlke, M.; Maher, T.J.; Ally, A. Modulation of cardiovascular responses and neurotransmission during peripheral nociception following nNOS antagonism within the periaqueductal gray. Brain Res. 2007, 1143, 150–160.
- Douki, T.; Cadet, J. Peroxynitrite mediated oxidation of purine bases of nucleosides and isolated DNA. Free Radic Res. 1996, 24, 369–380.
- McMahon, S.B. Wall and Melzack’s Textbook of Pain; Elsevier/Saunders Publishing: Philadelphia, PA, USA, 2013.
- Bakshi, N.; Morris, C.R. The role of the arginine metabolome in pain: Implications for sickle cell disease. J. Pain Res. 2016, 9, 167–175.
- Xanthos, D.N.; Bennett, G.J.; Coderre, T.J. Norepinephrine-induced nociception and vasoconstrictor hypersensitivity in rats with chronic post-ischemia pain. Pain 2008, 137, 640–651.
- Da Silva-Azevedo, L.; Baum, O.; Zakrzewicz, A.; Pries, A.R. Vascular endothelial growth factor is expressed in endothelial cells isolated from skeletal muscles of nitric oxide synthase knockout mice during prazosin-induced angiogenesis. Biochem. Biophys. Res. Commun. 2002, 297, 1270–1276.
- Bhattacharya, D.; Singh, M.K.; Chaudhuri, S.; Acharya, S.; Basu, A.K.; Chaudhuri, S. T11TS impedes glioma angiogenesis by inhibiting VEGF signaling and pro-survival PI3K/Akt/eNOS pathway with concomitant upregulation of PTEN in brain endothelial cells. J. Neurooncol. 2013, 113, 13–25.
- Chu, L.W.; Chen, J.Y.; Yu, K.L.; Cheng, K.I.; Wu, P.C.; Wu, B.N. Neuroprotective and anti-inflammatory activities of atorvastatin in a rat chronic constriction injury model. Int. J. Immunopathol. Pharmacol. 2012, 25, 219–230.
- Li, S.T.; Pan, J.; Hua, X.M.; Liu, H.; Shen, S.; Liu, J.F.; Li, B.; Tao, B.B.; Ge, X.L.; Wang, X.H.; et al. Endothelial nitric oxide synthase protects neurons against ischemic injury through regulation of brain-derived neurotrophic factor expression. CNS Neurosci. Ther. 2014, 20, 154–164.
- Huang, Y.; Jiao, B.; Zhu, B.; Xiong, B.; Lu, P.; Ai, L.; Yang, N.; Zhao, Y.; Xu, H. Nitric Oxide in the Spinal Cord Is Involved in the Hyperalgesia Induced by Tetrahydrobiopterin in Chronic Restraint Stress Rats. Front Neurosci. 2021, 15, 593654.
- Hu, Y.; Niu, X.; Wang, G.; Huang, J.; Liu, M.; Peng, B. Chronic prostatitis/chronic pelvic pain syndrome impairs erectile function through increased endothelial dysfunction, oxidative stress, apoptosis, and corporal fibrosis in a rat model. Andrology 2016, 4, 1209–1216.
- Han, T.H.; Hyduke, D.R.; Vaughn, M.W.; Fukuto, J.M.; Liao, J.C. Nitric oxide reaction with red blood cells and hemoglobin under heterogeneous conditions. Proc. Natl. Acad. Sci. USA 2002, 99, 7763–7768.
- Helms, C.; Kim-Shapiro, D.B. Hemoglobin-mediated nitric oxide signaling. Free Radic. Biol. Med. 2013, 61, 464–472.
- Boger, R.H. L-Arginine therapy in cardiovascular pathologies: Beneficial or dangerous? Curr. Opin. Clin. Nutr. Metab. Care 2008, 11, 55–61.
- Grimble, G.K. Adverse gastrointestinal effects of arginine and related amino acids. J. Nutr. 2007, 137 (Suppl. 2), 1693S–1701S.
- Rysz, J.; Franczyk, B.; Rysz-Gorzynska, M.; Gluba-Brzozka, A. Pharmacogenomics of Hypertension Treatment. Int. J. Mol. Sci. 2020, 21, 4709.
- Zhao, Q.; Zhang, J.; Wang, H. PGC-1alpha overexpression suppresses blood pressure elevation in DOCA-salt hypertensive mice. Biosci. Rep. 2015, 35, e00217.
- Du, Q.; Liao, Q.; Chen, C.; Yang, X.; Xie, R.; Xu, J. The Role of Transient Receptor Potential Vanilloid 1 in Common Diseases of the Digestive Tract and the Cardiovascular and Respiratory System. Front Physiol. 2019, 10, 1064.
- Randhawa, P.K.; Jaggi, A.S. TRPV1 channels in cardiovascular system: A double edged sword? Int. J. Cardiol. 2017, 228, 103–113.
- Ching, L.C.; Kou, Y.R.; Shyue, S.K.; Su, K.H.; Wei, J.; Cheng, L.C.; Yu, Y.B.; Pan, C.C.; Lee, T.S. Molecular mechanisms of activation of endothelial nitric oxide synthase mediated by transient receptor potential vanilloid type 1. Cardiovasc. Res. 2011, 91, 492–501.
- Xu, L.; Wang, S.; Li, B.; Sun, A.; Zou, Y.; Ge, J. A protective role of ciglitazone in ox-LDL-induced rat microvascular endothelial cells via modulating PPARgamma-dependent AMPK/eNOS pathway. J. Cell Mol. Med. 2015, 19, 92–102.
- Nunes, A.K.; Raposo, C.; Rocha, S.W.; Barbosa, K.P.; Luna, R.L.; da Cruz-Hofling, M.A.; Peixoto, C.A. Involvement of AMPK, IKbetaalpha-NFkappaB and eNOS in the sildenafil anti-inflammatory mechanism in a demyelination model. Brain Res. 2015, 1627, 119–133.
More
Information
Subjects:
Cardiac & Cardiovascular Systems
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
3.0K
Entry Collection:
Nitric Oxide: Physiology, Pharmacology, and Therapeutic Applications
Revisions:
2 times
(View History)
Update Date:
07 May 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No