 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Lidija Križančić Bombek | -- | 1843 | 2022-05-04 00:04:05 | | | |
| 2 | Jessie Wu | Meta information modification | 1843 | 2022-05-05 10:29:56 | | | | |
| 3 | Jessie Wu | Meta information modification | 1843 | 2022-05-05 10:49:45 | | |
Video Upload Options
Obesity and accompanying type 2 diabetes are among major and increasing worldwide problems that occur fundamentally due to excessive energy intake during its expenditure. Endotherms continuously consume a certain amount of energy to maintain core body temperature via thermogenic processes, mainly in brown adipose tissue and skeletal muscle. Skeletal muscle glucose utilization and heat production are significant and directly linked to body glucose homeostasis at rest, and especially during physical activity. However, this glucose balance is impaired in diabetic and obese states in humans and mice, and manifests as glucose resistance and altered muscle cell metabolism. Uncoupling proteins have a significant role in converting electrochemical energy into thermal energy without ATP generation. Different homologs of uncoupling proteins were identified, and their roles were linked to antioxidative activity and boosting glucose and lipid metabolism. From this perspective, uncoupling proteins were studied in correlation to the pathogenesis of diabetes and obesity and their possible treatments. Mice were extensively used as model organisms to study the physiology and pathophysiology of energy homeostasis. However, researchers should be aware of interstrain differences in mice models of obesity regarding thermogenesis and insulin resistance in skeletal muscles.
1. UCP Homologs and Their Roles
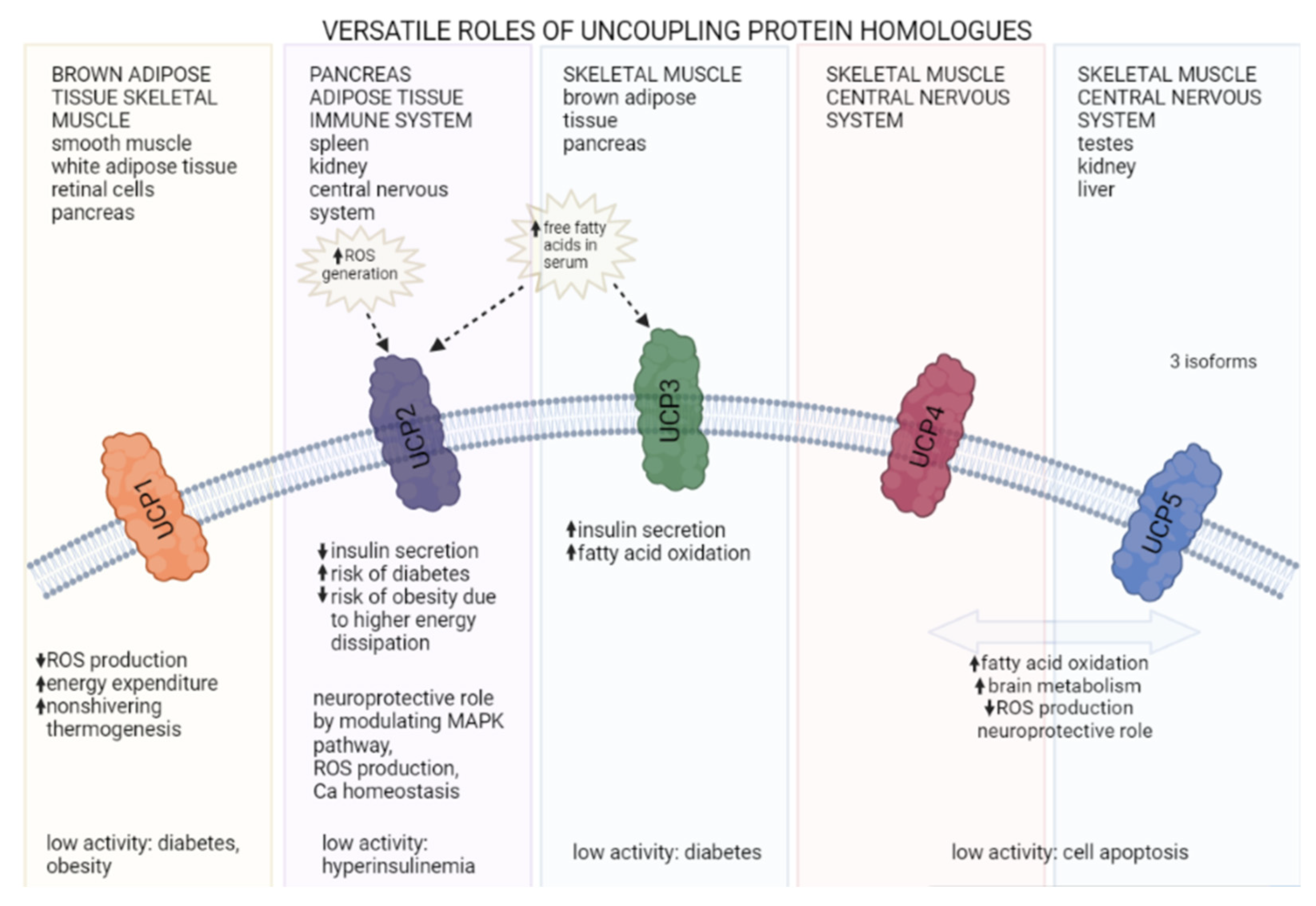
1.1. UCP1
1.2. UCP2
1.3. UCP3
1.4. Other UCPs
2. Sex Differences in UCP Expression
References
- Stefan Krauss; Chen-Yu Zhang; Bradford B. Lowell; The mitochondrial uncoupling-protein homologues. Nature Reviews Molecular Cell Biology 2005, 6, 248-261, 10.1038/nrm1592.
- Jing Liu; Ji Li; Wen-Jian Li; Chun-Ming Wang; The Role of Uncoupling Proteins in Diabetes Mellitus. Journal of Diabetes Research 2013, 2013, 1-7, 10.1155/2013/585897.
- Ryan J. Mailloux; Mary-Ellen Harper; Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radical Biology and Medicine 2011, 51, 1106-1115, 10.1016/j.freeradbiomed.2011.06.022.
- Petr Ježek; Blanka Holendová; Keith D. Garlid; Martin Jabůrek; Mitochondrial Uncoupling Proteins: Subtle Regulators of Cellular Redox Signaling. Antioxidants & Redox Signaling 2018, 29, 667-714, 10.1089/ars.2017.7225.
- Barbara Cannon; Jan Nedergaard; Brown Adipose Tissue: Function and Physiological Significance. Physiological Reviews 2004, 84, 277-359, 10.1152/physrev.00015.2003.
- Telma Cristina Esteves; Martin D. Brand; The reactions catalysed by the mitochondrial uncoupling proteins UCP2 and UCP3. Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms 2005, 1709, 35-44, 10.1016/j.bbabio.2005.06.002.
- Marta Giralt; Immaculada Martin; Roser Iglesias; Octavi Vinas; Francesc Villarroya; Teresa Mampel; Ontogeny and perinatal modulation of gene expression in rat brown adipose tissue. Unaltered iodothyronine 5'-deiodinase activity is necessary for the response to environmental temperature at birth. JBIC Journal of Biological Inorganic Chemistry 1990, 193, 297-302, 10.1111/j.1432-1033.1990.tb19336.x.
- Josef Houštěk; Jan Kopecky; Zdeněk Rychter; Tomáš Soukup; Uncoupling protein in embryonic brown adipose tissue — existence of nonthermogenic and thermogenic mitochondria. Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms 1988, 935, 19-25, 10.1016/0005-2728(88)90103-x.
- Abhijit Babaji Shinde; Anying Song; Qiong A. Wang; Brown Adipose Tissue Heterogeneity, Energy Metabolism, and Beyond. Frontiers in Endocrinology 2021, 12, 651763, 10.3389/fendo.2021.651763.
- Anying Song; Wenting Dai; Min Jee Jang; Leonard Medrano; Zhuo Li; Hu Zhao; Mengle Shao; Jiayi Tan; Aimin Li; Tinglu Ning; et al.Marcia M. MillerBrian ArmstrongJanice M. HussYi ZhuYong LiuViviana GradinaruXiwei WuLei JiangPhilipp E. SchererQiong A. Wang Low- and high-thermogenic brown adipocyte subpopulations coexist in murine adipose tissue. Journal of Clinical Investigation 2019, 130, 247-257, 10.1172/jci129167.
- L. J. Bukowiecki; A. Géloën; A. J. Collet; Proliferation and differentiation of brown adipocytes from interstitial cells during cold acclimation. American Journal of Physiology-Cell Physiology 1986, 250, C880-C887, 10.1152/ajpcell.1986.250.6.c880.
- Yun‐Hee Lee; Anelia P. Petkova; Anish A. Konkar; James G. Granneman; Cellular origins of cold-induced brown adipocytes in adult mice. The FASEB Journal 2014, 29, 286-299, 10.1096/fj.14-263038.
- Andriy Fedorenko; Polina V. Lishko; Yuriy Kirichok; Mechanism of Fatty-Acid-Dependent UCP1 Uncoupling in Brown Fat Mitochondria. Cell 2012, 151, 400-413, 10.1016/j.cell.2012.09.010.
- R Burcelin; J Kande; D Ricquier; J Girard; Changes in uncoupling protein and GLUT4 glucose transporter expressions in interscapular brown adipose tissue of diabetic rats: relative roles of hyperglycaemia and hypoinsulinaemia. Biochemical Journal 1993, 291, 109-113, 10.1042/bj2910109.
- Matthias J. Betz; Sven Enerbäck; Targeting thermogenesis in brown fat and muscle to treat obesity and metabolic disease. Nature Reviews Endocrinology 2017, 14, 77-87, 10.1038/nrendo.2017.132.
- Michèle M Sale; Fang-Chi Hsu; Nicholette D Palmer; Candace J Gordon; Keith L Keene; Hermina M Borgerink; Arun J Sharma; Richard N Bergman; Kent D Taylor; Mohammed F Saad; et al.Jill M Norris The uncoupling protein 1 gene, UCP1, is expressed in mammalian islet cells and associated with acute insulin response to glucose in African American families from the IRAS Family Study. BMC Endocrine Disorders 2007, 7, 1-1, 10.1186/1472-6823-7-1.
- Adjeitey CN, Mailloux RJ, Dekemp RA, Harper ME.; Mitochondrial uncoupling in skeletal muscle by UCP1 augments energy expenditure and glutathione content while mitigating ROS production.. Am J Physiol Endocrinol Metab 2013, 305(3), E405-E415, doi:10.1152/ajpendo.00057.2013.
- ite Gaudry MJ, Campbell KL, Jastroch M; Evolution of UCP1. Handb Exp Pharmacol 2019, 251, 127-141, doi:10.1007/164_2018_116.
- Janne Orava; Pirjo Nuutila; Martin E. Lidell; Vesa Oikonen; Tommi Noponen; Tapio Viljanen; Mika Scheinin; Markku Taittonen; Tarja Niemi; Sven Enerbäck; et al.Kirsi A. Virtanen Different Metabolic Responses of Human Brown Adipose Tissue to Activation by Cold and Insulin. Cell Metabolism 2011, 14, 272-279, 10.1016/j.cmet.2011.06.012.
- Edward T. Chouchani; Lawrence Kazak; Bruce M. Spiegelman; New Advances in Adaptive Thermogenesis: UCP1 and Beyond. Cell Metabolism 2018, 29, 27-37, 10.1016/j.cmet.2018.11.002.
- Jun-Jing Jia; Yun-Bo Tian; Zhen-Hui Cao; Lin-Li Tao; Xi Zhang; Si-Zhen Gao; Chang-Rong Ge; Qiu-Ye Lin; M. Jois; The polymorphisms of UCP1 genes associated with fat metabolism, obesity and diabetes. Molecular Biology Reports 2009, 37, 1513-1522, 10.1007/s11033-009-9550-2.
- A Hamann; J Tafel; B Büsing; H Münzberg; A Hinney; H Mayer; W Siegfried; D Ricquier; H Greten; J Hebebrand; et al.S Matthaei Analysis of the uncoupling protein-1 (UCP1) gene in obese and lean subjects: Identification of four amino acid variants. International Journal of Obesity 1998, 22, 939-941, 10.1038/sj.ijo.0800725.
- Kil Soo Kim; Dae-Yeon Cho; Young Joo Kim; Sun Mi Choi; Jong Yeol Kim; Seung Uoo Shin; Yoo Sik Yoon; The finding of new genetic polymorphism of UCP-1 A-1766G and its effects on body fat accumulation. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2005, 1741, 149-155, 10.1016/j.bbadis.2004.11.026.
- S Brun; M C Carmona; T Mampel; O Viñas; M Giralt; R Iglesias; F Villarroya; Uncoupling protein-3 gene expression in skeletal muscle during development is regulated by nutritional factors that alter circulating non-esterified fatty acids.. FEBS Letters 1999, 453, 205-209, 10.1016/s0014-5793(99)00722-x.
- Zhang CY, Baffy G, Perret P, Krauss S, Peroni O, Grujic D, Hagen T, Vidal-Puig AJ, Boss O, Kim YB, Zheng XX, Wheeler MB, Shulman GI, Chan CB, Lowell BB; Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell 2001, 105(6), 745-755, doi:10.1016/s0092-8674(01)00378-6.
- Stefan Krauss; Chen-Yu Zhang; Luca Scorrano; Louise T. Dalgaard; Julie St-Pierre; Shane T. Grey; Bradford B. Lowell; Superoxide-mediated activation of uncoupling protein 2 causes pancreatic β cell dysfunction. Journal of Clinical Investigation 2003, 112, 1831-1842, 10.1172/jci19774.
- Zhongmin Alex Ma; Zhengshan Zhao; John Turk; Mitochondrial Dysfunction and β-Cell Failure in Type 2 Diabetes Mellitus. Experimental Diabetes Research 2011, 2012, 1-11, 10.1155/2012/703538.
- Cláudio T. De Souza; Eliana P. Araújo; Luiz F. Stoppiglia; José R. Pauli; Eduardo Ropelle; Silvana A. Rocco; Rodrigo M. Marin; Kleber G. Franchini; José B. Carvalheira; Mário J. Saad; et al.Antonio C. BoscheroEverardo M. CarneiroLício A. Velloso Inhibition of UCP2 expression reverses diet-induced diabetes mellitus by effects on both insulin secretion and action. The FASEB Journal 2007, 21, 1153-1163, 10.1096/fj.06-7148com.
- Zhang CY, Baffy G, Perret P, Krauss S, Peroni O, Grujic D, Hagen T, Vidal-Puig AJ, Boss O, Kim YB, Zheng XX, Wheeler MB, Shulman GI, Chan CB, Lowell BB; Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell 2001, 105(6), 745-55, doi: 10.1016/s0092-8674(01)00378-6.
- Christine A. Robson-Doucette; Sobia Sultan; Emma M. Allister; Jakob D. Wikstrom; Vasilij Koshkin; Alpana Bhattacharjee; Kacey J. Prentice; Samuel B. Sereda; Orian S. Shirihai; Michael B. Wheeler; et al. Beta-Cell Uncoupling Protein 2 Regulates Reactive Oxygen Species Production, Which Influences Both Insulin and Glucagon Secretion. Diabetes 2011, 60, 2710-2719, 10.2337/db11-0132.
- Rong Huang; Tingting Cai; Yunting Zhou; Yuming Wang; Huiying Wang; Ziyang Shen; Wenqing Xia; Xiaomei Liu; Bo Ding; Yong Luo; et al.Rengna YanHuiqin LiJindan WuJianhua Ma Ethnicity Differences in the Association of UCP1-3826A/G, UCP2-866G/A and Ala55Val, and UCP3-55C/T Polymorphisms with Type 2 Diabetes Mellitus Susceptibility: An Updated Meta-Analysis. BioMed Research International 2021, 2021, 1-14, 10.1155/2021/3482879.
- Yalin Emre; Tobias Nübel; Uncoupling protein UCP2: When mitochondrial activity meets immunity. FEBS Letters 2010, 584, 1437-1442, 10.1016/j.febslet.2010.03.014.
- Sabrina Diano; Tamas L. Horvath; Mitochondrial uncoupling protein 2 (UCP2) in glucose and lipid metabolism. Trends in Molecular Medicine 2012, 18, 52-58, 10.1016/j.molmed.2011.08.003.
- P Andy Li Suresh L Mehta; P. Andy Li; Neuroprotective Role of Mitochondrial Uncoupling Protein 2 in Cerebral Stroke. British Journal of Pharmacology 2009, 29, 1069-1078, 10.1038/jcbfm.2009.4.
- Olivier Boss; Sonia Samec; Ariane Paoloni-Giacobino; Colette Rossier; Abdul Dulloo; Josiane Seydoux; Patrick Muzzin; Jean-Paul Giacobino; Uncoupling protein-3: a new member of the mitochondrial carrier family with tissue-specific expression. FEBS Letters 1997, 408, 39-42, 10.1016/s0014-5793(97)00384-0.
- Antonio Vidal-Puig; Gemma Solanes; Danica Grujic; Jeffrey S. Flier; Bradford B. Lowell; UCP3: An Uncoupling Protein Homologue Expressed Preferentially and Abundantly in Skeletal Muscle and Brown Adipose Tissue. Biochemical and Biophysical Research Communications 1997, 235, 79-82, 10.1006/bbrc.1997.6740.
- Da-Wei Gong; Yufang He; Michael Karas; Marc Reitman; Uncoupling Protein-3 Is a Mediator of Thermogenesis Regulated by Thyroid Hormone, beta3-Adrenergic Agonists, and Leptin. Journal of Biological Chemistry 1997, 272, 24129-24132, 10.1074/jbc.272.39.24129.
- Antonio Vidal-Puig; Danica Grujic; Chen-Yu Zhang; Thilo Hagen; Olivier Boss; Yasuo Ido; Alicja Szczepanik; Jennifer Wade; Vamsi Mootha; Ronald Cortright; et al.Deborah MuoioBradford B. Lowell Energy Metabolism in Uncoupling Protein 3 Gene Knockout Mice. Journal of Biological Chemistry 2000, 275, 16258-16266, 10.1074/jbc.m910179199.
- Karolina E. Hilse; Anastasia V. Kalinovich; Anne Rupprecht; Alina Smorodchenko; Ute Zeitz; Katrin Staniek; Reinhold G. Erben; Elena E. Pohl; The expression of UCP3 directly correlates to UCP1 abundance in brown adipose tissue. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2015, 1857, 72-78, 10.1016/j.bbabio.2015.10.011.
- Vian Azzu; Martin Jastroch; Ajit S. Divakaruni; Martin D. Brand; The regulation and turnover of mitochondrial uncoupling proteins. Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms 2010, 1797, 785-791, 10.1016/j.bbabio.2010.02.035.
- Lukáš Alán; Katarína Smolková; Eva Kronusová; Jitka Šantorová; Petr Ježek; Absolute levels of transcripts for mitochondrial uncoupling proteins UCP2, UCP3, UCP4, and UCP5 show different patterns in rat and mice tissues. Journal of Bioenergetics and Biomembranes 2009, 41, 71-78, 10.1007/s10863-009-9201-2.
- D. S. Weigle; L. E. Selfridge; M. W. Schwartz; R. J. Seeley; D. E. Cummings; P. J. Havel; J. L. Kuijper; H. BeltrandelRio; Elevated free fatty acids induce uncoupling protein 3 expression in muscle: a potential explanation for the effect of fasting. Diabetes 1998, 47, 298-302, 10.2337/diabetes.47.2.298.
- N Pedraza; G Solanes; M C Carmona; R Iglesias; O Viñas; T Mampel; M Vazquez; M Giralt; F Villarroya; Impaired expression of the uncoupling protein-3 gene in skeletal muscle during lactation: fibrates and troglitazone reverse lactation-induced downregulation of the uncoupling protein-3 gene.. Diabetes 2000, 49, 1224-1230, 10.2337/diabetes.49.7.1224.
- Yunfeng Li; Kathrin Maedler; Luan Shu; Leena Haataja; UCP-2 and UCP-3 Proteins Are Differentially Regulated in Pancreatic Beta-Cells. PLoS ONE 2008, 3, e1397, 10.1371/journal.pone.0001397.
- Sheila R. Costford; Shehla N. Chaudhry; Sean A. Crawford; Mahmoud Salkhordeh; Mary-Ellen Harper; Long-term high-fat feeding induces greater fat storage in mice lacking UCP3. American Journal of Physiology-Endocrinology and Metabolism 2008, 295, E1018-E1024, 10.1152/ajpendo.00779.2007.
- Graham P. Holloway; Swati S. Jain; Veronic Bezaire; Xiao Xia Han; Jan F. C. Glatz; Joost J. F. P. Luiken; Mary-Ellen Harper; Arend Bonen; FAT/CD36-null mice reveal that mitochondrial FAT/CD36 is required to upregulate mitochondrial fatty acid oxidation in contracting muscle. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology 2009, 297, R960-R967, 10.1152/ajpregu.91021.2008.
- Patrick Schrauwen; Marco Mensink; Gert Schaart; Esther Moonen-Kornips; Jean-Pierre Sels; Ellen E. Blaak; Aaron P. Russell; Matthijs K. C. Hesselink; Reduced Skeletal Muscle Uncoupling Protein-3 Content in Prediabetic Subjects and Type 2 Diabetic Patients: Restoration by Rosiglitazone Treatment. The Journal of Clinical Endocrinology & Metabolism 2006, 91, 1520-1525, 10.1210/jc.2005-1572.
- Miranda Nabben; Joris Hoeks; Mitochondrial uncoupling protein 3 and its role in cardiac- and skeletal muscle metabolism. Physiology & Behavior 2008, 94, 259-269, 10.1016/j.physbeh.2007.11.039.
- Vian Azzu; Martin D. Brand; The on-off switches of the mitochondrial uncoupling proteins. Trends in Biochemical Sciences 2010, 35, 298-307, 10.1016/j.tibs.2009.11.001.
- Sophie Rousset; Julien Mozo; Geneviève Dujardin; Yalin Emre; Sandrine Masscheleyn; Daniel Ricquier; Anne-Marie Cassard-Doulcier; UCP2 is a mitochondrial transporter with an unusual very short half-life. FEBS Letters 2007, 581, 479-482, 10.1016/j.febslet.2007.01.010.
- P Puigserver; David Herron; M Gianotti; A Palou; Barbara Cannon; Jan Nedergaard; Induction and degradation of the uncoupling protein thermogenin in brown adipocytes in vitro and in vivo. Evidence for a rapidly degradable pool. Biochemical Journal 1992, 284, 393-398, 10.1042/bj2840393.
- Claire Pecqueur; Marie-Clotilde Alves-Guerra; Chantal Gelly; Corinne Lévi-Meyrueis; Elodie Couplan; Sheila Collins; Daniel Ricquier; Frederic Bouillaud; Bruno Miroux; Uncoupling Protein 2, in Vivo Distribution, Induction upon Oxidative Stress, and Evidence for Translational Regulation. Journal of Biological Chemistry 2001, 276, 8705-8712, 10.1074/jbc.m006938200.
- Daniel Sanchis; Christophe Fleury; Nathalie Chomiki; Marc Goubern; Quinling Huang; Maria Neverova; Francine Grégoire; Juliet Easlick; Serge Raimbault; Corinne Lévi-Meyrueis; et al.Bruno MirouxSheila CollinsMichael SeldinDenis RichardCraig WardenFrédéric BouillaudDaniel Ricquier BMCP1, a Novel Mitochondrial Carrier with High Expression in the Central Nervous System of Humans and Rodents, and Respiration Uncoupling Activity in Recombinant Yeast. Journal of Biological Chemistry 1998, 273, 34611-34615, 10.1074/jbc.273.51.34611.
- Weiguang Mao; Xing Xian Yu; Alan Zhong; Wenlu Li; Jennifer Brush; Steven W Sherwood; Sean H Adams; Guohua Pan; UCP4, a novel brain-specific mitochondrial protein that reduces membrane potential in mammalian cells. FEBS Letters 1999, 443, 326-330, 10.1016/s0014-5793(98)01713-x.
- Xiaolin Yang; Richard E. Pratley; Stephen Tokraks; P. Antonio Tataranni; Paska A. Permana; UCP5/BMCP1 transcript isoforms in human skeletal muscle: relationship of the short-insert isoform with lipid oxidation and resting metabolic rates. Molecular Genetics and Metabolism 2002, 75, 369-373, 10.1016/s1096-7192(02)00008-2.
- David B. Ramsden; Philip W.‐L. Ho; Jessica W.‐M. Ho; Hui‐Fang Liu; Danny H.‐F. So; Ho‐Man Tse; Koon‐Ho Chan; Shu‐Leong Ho; Human neuronal uncoupling proteins 4 and 5 (UCP4 and UCP5): structural properties, regulation, and physiological role in protection against oxidative stress and mitochondrial dysfunction. Brain and Behavior 2012, 2, 468-478, 10.1002/brb3.55.
- A M Rodríguez; A Palou; Uncoupling proteins: gender dependence and their relation to body weight control. International Journal of Obesity 2004, 28, 500-502, 10.1038/sj.ijo.0802588.
- Shao-Hua Yang; Ran Liu; Evelyn J. Perez; Yi Wen; Stanley M. Stevens; Thomas Valencia; Anne-Marie Brun-Zinkernagel; Laszlo Prokai; Yvonne Will; James Dykens; et al.Peter KoulenJames W. Simpkins Mitochondrial localization of estrogen receptor beta. Proceedings of the National Academy of Sciences 2004, 101, 4130-4135, 10.1073/pnas.0306948101.
- Jane M. Eason; Gail A. Schwartz; Grace K. Pavlath; Arthur W. English; Sexually dimorphic expression of myosin heavy chains in the adult mouse masseter. Journal of Applied Physiology 2000, 89, 251-258, 10.1152/jappl.2000.89.1.251.
- A. M. Rodríguez; M. Monjo; P. Roca; A. Palou; Opposite actions of testosterone and progesterone on UCP1 mRNA expression in cultured brown adipocytes. Cellular and Molecular Life Sciences 2002, 59, 1714-1723, 10.1007/pl00012499.
- Adamo Valle; Francisco García-Palmer; Jordi Oliver Oliver; Pilar Roca; Sex Differences in Brown Adipose Tissue Thermogenic Features During Caloric Restriction. Cellular Physiology and Biochemistry 2007, 19, 195-204, 10.1159/000099207.
- Michael Moschinger; Karolina E. Hilse; Anne Rupprecht; Ute Zeitz; Reinhold G. Erben; Thomas Rülicke; Elena E. Pohl; Age-related sex differences in the expression of important disease-linked mitochondrial proteins in mice. Biology of Sex Differences 2019, 10, 1-10, 10.1186/s13293-019-0267-1.
- Céline Aguer; Oliver Fiehn; Erin L. Seifert; Véronic Bézaire; John K. Meissen; Amanda Daniels; Kyle Scott; Jean‐Marc Renaud; Marta Padilla; David R. Bickel; et al.Michael DysartSean H. AdamsMary‐Ellen Harper Muscle uncoupling protein 3 overexpression mimics endurance training and reduces circulating biomarkers of incomplete β‐oxidation. The FASEB Journal 2013, 27, 4213-4225, 10.1096/fj.13-234302.
- A M Rodríguez; A Palou; Uncoupling proteins: gender-dependence and their relation to body weight control. International Journal of Obesity 2004, 28, 327-329, 10.1038/sj.ijo.0802579.
- Nadezhda Bazhan; Tatiana Jakovleva; Natalia Feofanova; Elena Denisova; Anastasia Dubinina; Natalia Sitnikova; Elena Makarova; Sex Differences in Liver, Adipose Tissue, and Muscle Transcriptional Response to Fasting and Refeeding in Mice. Cells 2019, 8, 1529, 10.3390/cells8121529.
- A M Rodríguez; A Palou; Uncoupling proteins: gender-dependence and their relation to body weight control. International Journal of Obesity 2004, 28, 327-329, 10.1038/sj.ijo.0802579.
- Nadezhda Bazhan; Tatiana Jakovleva; Natalia Feofanova; Elena Denisova; Anastasia Dubinina; Natalia Sitnikova; Elena Makarova; Sex Differences in Liver, Adipose Tissue, and Muscle Transcriptional Response to Fasting and Refeeding in Mice. Cells 2019, 8, 1529, 10.3390/cells8121529.


