Proteins are the workhorses of the cell. With different combinations of the 20 common amino acids and some modifications of these amino acids, proteins have evolved with a staggering array of new functions and capabilities due to Protein Engineering techniques. The practical course presented was offered to undergraduate bioengineering and chemical students at the Faculty of Engineering of the University of Porto (Portugal) and consists of sequential laboratory sessions to learn the basic skills related to the expression and purification of recombinant proteins in bacterial hosts. These experiments were successfully applied by students as all working groups were able to isolate a model recombinant protein (the enhanced green fluorescent protein, eGFP) from a cell lysate containing a mixture of proteins and other biomolecules produced by an Escherichia coli strain and evaluate the performance of the extraction and purification procedures they learned.
1. Introduction
The engineering of proteins represents a modern and powerful approach to generating novel proteins for applications in different fields such as biocatalysts, therapeutic agents, and biosensors
[1]. Therefore, knowledge of the basic skills of Protein Engineering is mandatory for future bioengineers and chemical engineers specialized in Biotechnology.
Green fluorescent protein (GFP) is a small protein of about 27 kDa consisting of 238 amino acids (aa) derived from the jellyfish
Aequorea victoria [2]. It is intrinsically fluorescent, emitting a brilliant green light when exposed to ultraviolet or blue light, due to a chromophore formed from a maturation reaction of three specific aa at the center of the protein (Ser65, Tyr66, and Gly67)
[3][4].
GFP-like proteins are widely used as quantitative genetically encoded markers for studying protein-protein interactions and cell tracking
[5][6]. One of the most interesting aspects of GFP over other fluorescent tags is that the chromophore forms spontaneously and without accessory co-factors, substrates, or enzymes; it only requires the presence of oxygen during maturation
[7], which means that the gene could be taken directly from
A. victoria and expressed in other organisms as the Gram-negative bacterium
Escherichia coli while still maintaining fluorescence.
The heterologous expression of GFP is a particularly interesting system for didactic purposes since it can be easily observed during laboratory classes. To this end,
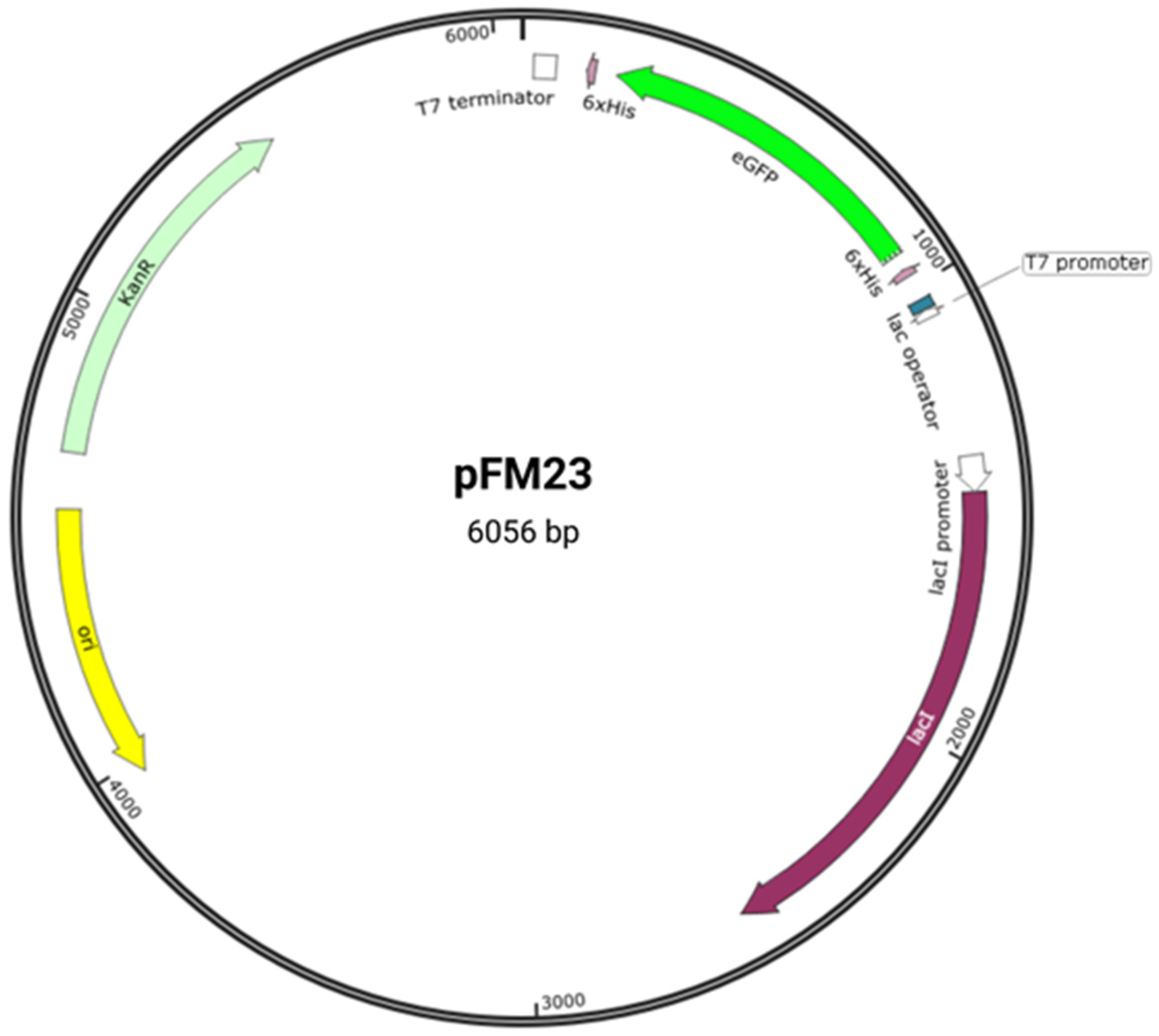
scholars previously cloned the eGFP gene fused to histidine (His) tags in the pET28a vector
[8], generating plasmid pFM23 (
Figure 1). Plasmid pFM23 for cytoplasmic production of eGFP-His6 was constructed by digestion of plasmid pFM20 (expressing ZZ-GFP) with the NdeI/BamHI restriction enzymes and cloning of the eGFP gene into pET28A
[8]. For insertion into plasmid pFM20, the eGFP coding sequence had previously been amplified from plasmid pEGFP-N1
[8]. Expression using a pET-based vector such as pET28a provides larger amounts of the target protein than other simplified systems. For this system,
E. coli host cells engineered to carry the gene encoding T7 RNA polymerase downstream of the lac promoter are required. These cells are transformed with a plasmid that includes a copy of the T7 promoter and, adjacent to it, the gene to be expressed.
Figure 1. Plasmid pFM23 map. This harbours (i) a pMB1 origin of replication (ori), (ii) a repressor for the lac promoter (lacI), (iii) a transcriptional promoter from the T7 phage (T7 promoter), (iv) a lactose operator (lac operator), (v) an affinity purification tag (6 × His), (vi) a T7 transcriptional terminator (T7 terminator), (vii) a kanamycin resistance gene (KanR), and (viii) the eGFP gene (eGFP).
When IPTG, a lactose analog, is added to the culture medium, T7 RNA polymerase is expressed by transcription from the lac promoter
[9]. The enzyme recognizes the T7 promoter on the plasmid and catalyzes the transcription of the gene of interest. T7 RNA polymerase is so selective and active that almost all of the cell resources are directed to recombinant protein expression
[10]. The bacterium
E. coli is a preferred host for the production of recombinant proteins
[11][12] due to its fast growth at high cell densities, minimal nutrient requirements, well-known genetics, and availability of a large number of cloning vectors and mutant host strains
[13]. This bacterium can accumulate many recombinant proteins to at least 20% of the total cell protein content
[14] and translocate them from the cytoplasm to the periplasm
[15]. Despite all these advantages, the expression of recombinant proteins using
E. coli as host often results in the formation of insoluble protein aggregates called inclusion bodies
[11][16]. Inclusion bodies are usually formed in the cytoplasm, and several methods have been described for the redirection of proteins from inclusion bodies into the soluble cytoplasmic fraction of cells
[17]. Overall, they can be divided into procedures where protein is refolded from inclusion bodies and procedures where the expression strategy is modified to obtain soluble proteins by lowering the expression levels. For instance, this can be achieved by balancing the promoter strength and gene copy number
[15][18].
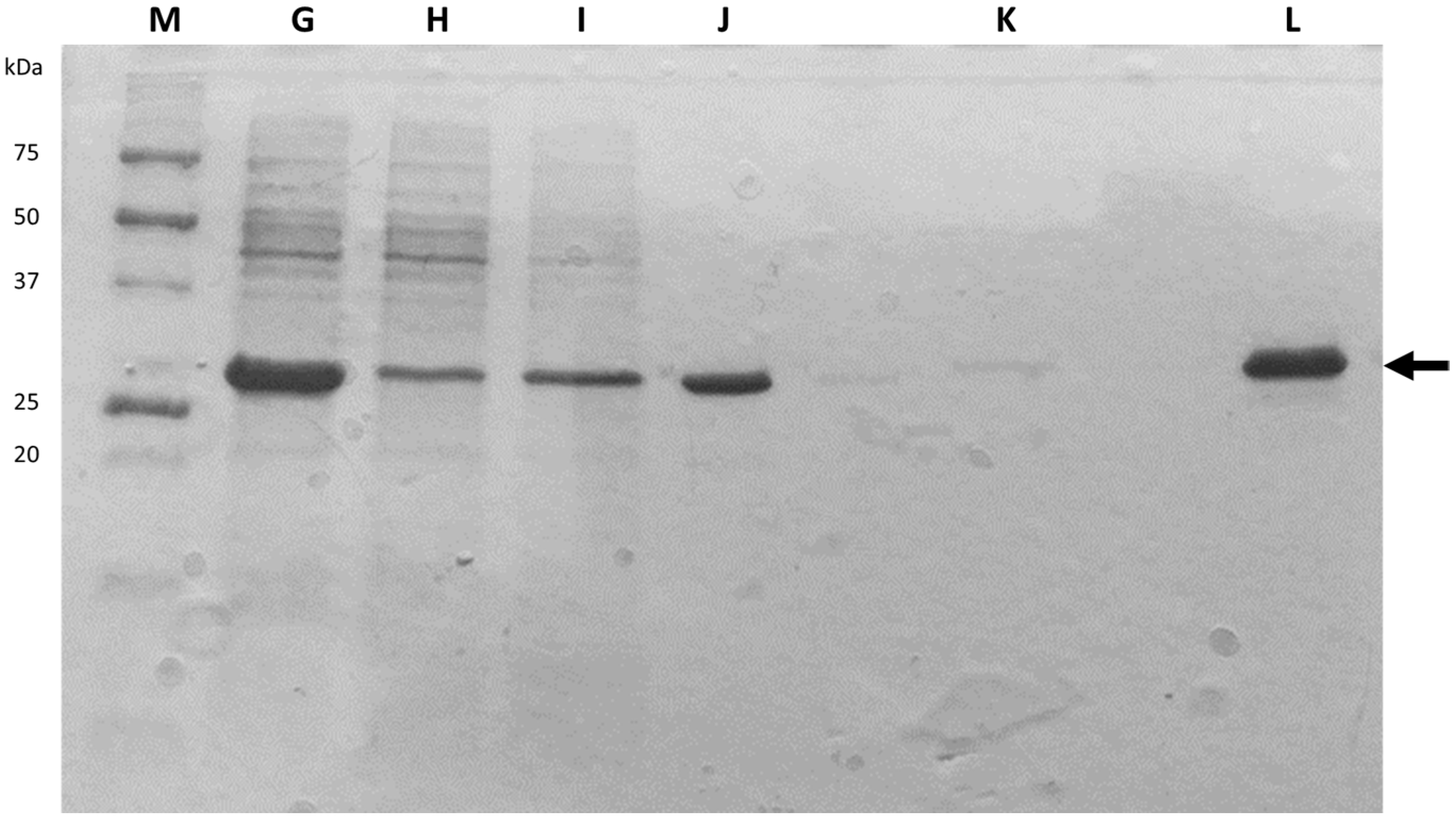
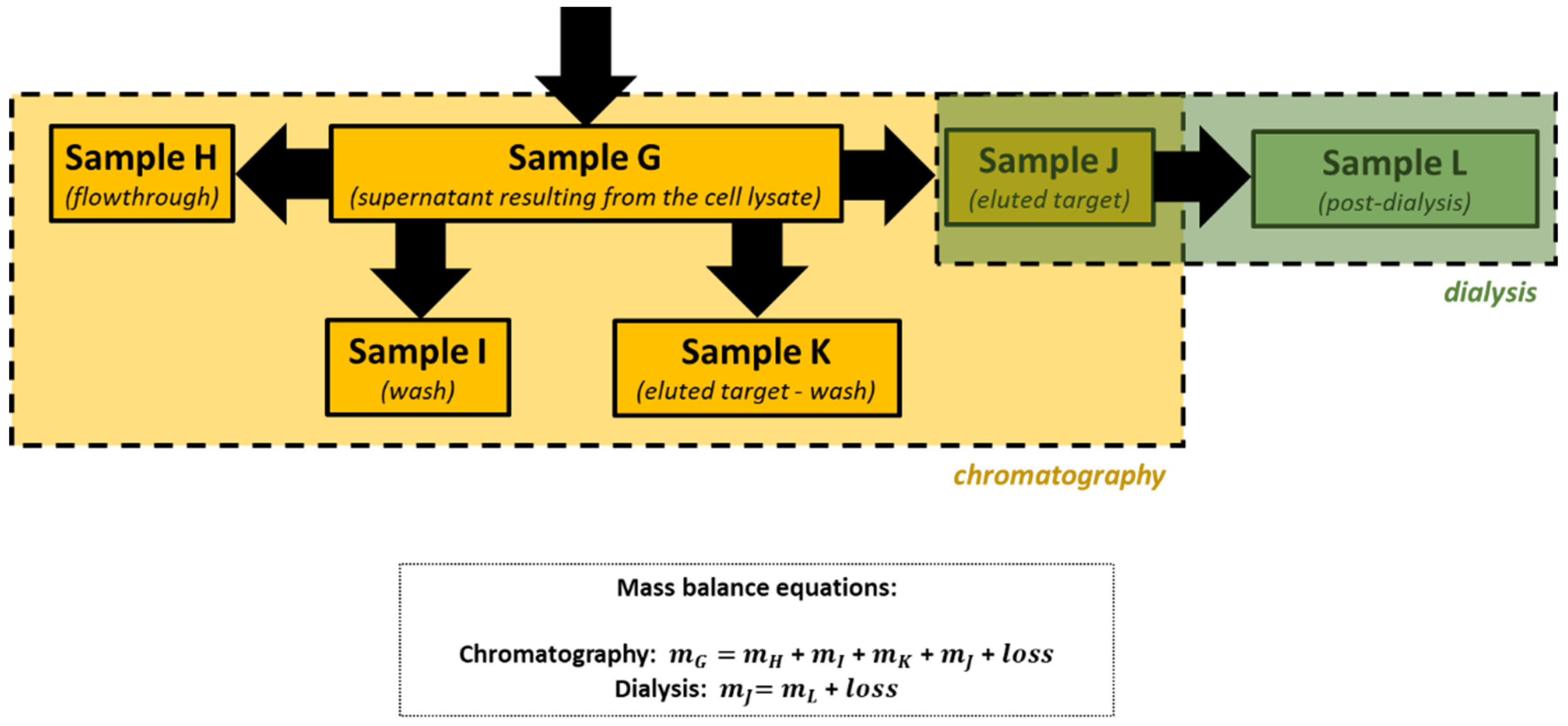
After cellular disruption, several methods can be used to enrich or purify a protein of interest from other proteins and components in a crude cell lysate. One of the most powerful methods is affinity chromatography, whereby the protein of interest is purified by its specific binding properties to an immobilized ligand
[19]. In this practical course, protein purification was performed by affinity chromatography of the His-tagged protein in a nickel column, followed by dialysis. His-tag expression systems are extensively used in Protein Engineering because His-tagged proteins can be easily purified by single-step affinity chromatography, namely immobilized metal affinity chromatography (IMAC), which is commercially available in different kinds of formats, the Ni-NTA matrices being the most widely used
[20]. Moreover, His-tags have low molecular weight (∼2.5 kDa) and usually do not affect protein structure and function, which means that it is not necessary to separate the His-tag from the target protein
[21]. Most other proteins in the lysate do not bind to the Ni-NTA resin, or bind only weakly, thus the use of His-tag and IMAC can provide relatively pure recombinant protein directly from a crude lysate.
2. Pedagogical Considerations
The practical course is offered to undergraduate bioengineering and chemical engineering students at the Faculty of Engineering of the University of Porto (Portugal). A prerequisite for attendance is basic knowledge of cellular biology, molecular biology, and microbiology.
Students should familiar with the fundamentals of DNA cloning, vector design, and the pET system, since these concepts were attained in the corresponding lecture courses. For that reason, no pre-lab lecture was given, and students were expected to understand the lab work with the support of raw protocols provided by the instructors. Before starting the experimental session, a working group was selected to make a brief presentation of the theoretical concepts related to the topic of the session, as well as to present a quick protocol that was distributed to the remaining groups. Doubts were clarified and the quick protocols prepared by the remaining groups were collected. At the end of the course, the groups had access to the raw data of the other groups from the class and treated these data as a whole in the writing of their reports.
The main student learning objectives were:
-
To be proficient in carrying out the following procedures: bacterial growth, cell lysis, protein purification, protein quantification, and polyacrylamide gel electrophoresis.
-
To reinforce understanding of the following topics: plasmid design, recombinant protein expression, and protein purification.
-
To acquire skills to operate the following equipment: UV-Vis spectrophotometer, centrifuges, sonicator, microplate reader, and electrophoresis apparatus.
-
To improve the ability in critical thinking, team organization, and scientific concepts exposition and writing skills.
3. A Practical Teaching Course of Protein Engineering
3.1. Bacterial Growth
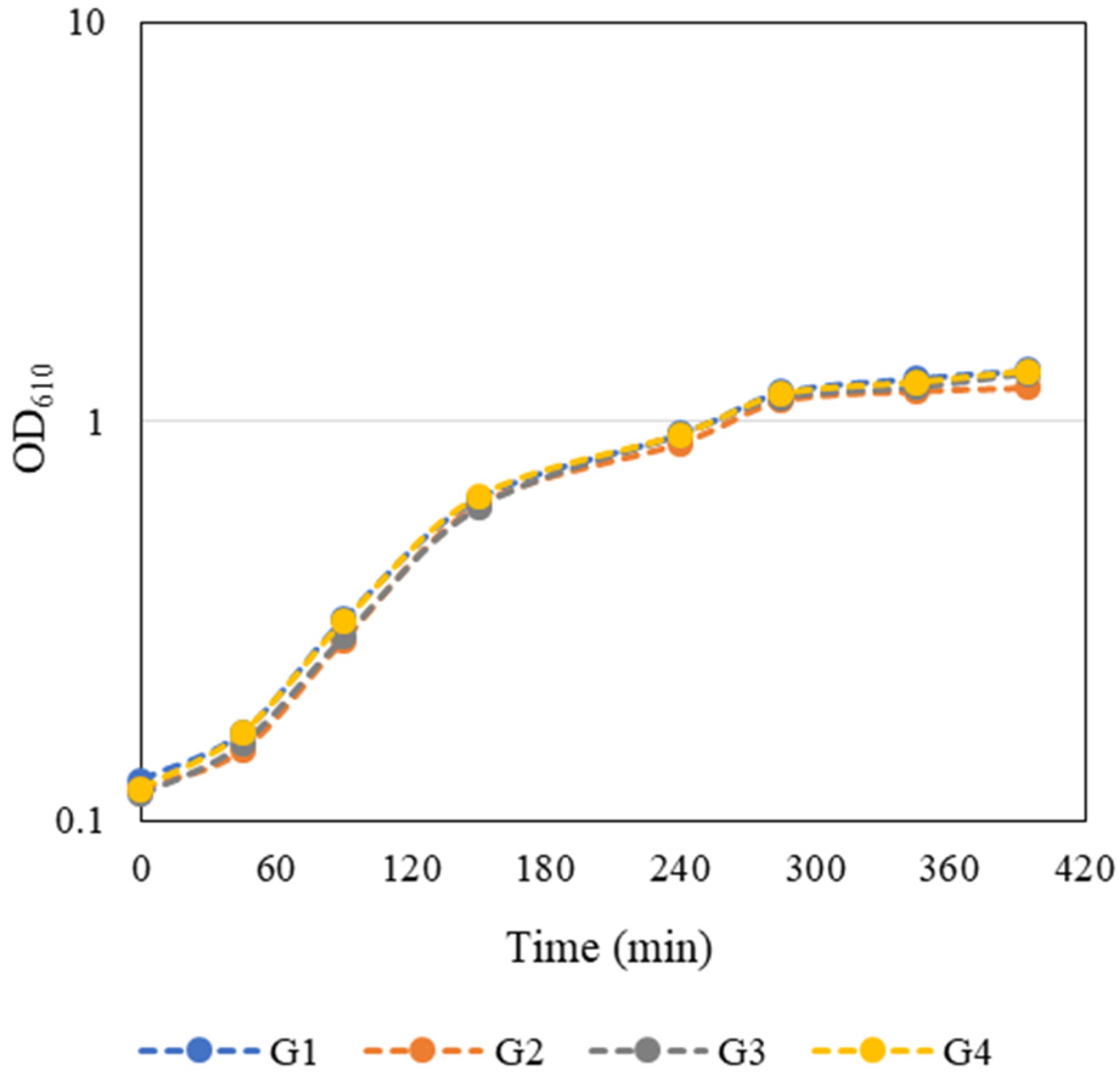
An overnight culture of E. coli JM109(DE3) containing the pFM23 plasmid was given to each group. To start the bacterial growth curves, the optical density at 610 nm (OD610) of this stationary phase culture was determined, and a dilution factor was calculated such that by adding fresh 125 mL of LB medium the final OD610 would be approximately 0.1. E. coli growth curves presented in Figure 2 were constructed by measuring the OD610 every 45–60 min during class and in the first hours after class. The growth curves were very similar and the groups considered that the exponential phase of bacterial growth occurred between 90 and 285 min. Growth kinetics parameters such as maximum growth rate (µmax) and doubling time (td) (Table 1) were then calculated separately for each individual growth curve through the logarithmic representation of the exponential part of the growth curve. Regression analysis of this experimental data was performed using a Microsoft Excel spreadsheet. The slope of the line that best fits the points corresponds to µmax of each independent growth, whereas td was estimated by Equation (1):
Figure 2. Student-generated growth curves of E. coli JM109(DE3) harbouring the pFM23 plasmid. After 180 min of incubation, IPTG was added to the culture medium.
Table 1. Parameters obtained by regression analysis for each group.
| Group |
R2 * |
p-Value ** |
µmax (min−1) |
td (min) |
| G1 |
0.9430 |
0.0289 |
0.00634 |
109.4 |
| G2 |
0.9270 |
0.0372 |
0.00660 |
105.0 |
| G3 |
0.9401 |
0.0304 |
0.00673 |
103.0 |
| G4 |
0.9349 |
0.0331 |
0.00631 |
109.8 |
 +1 credit
+1 credit