 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Anna Gharibyan | -- | 3744 | 2022-04-28 13:29:01 | | | |
| 2 | Jessie Wu | -3 word(s) | 3741 | 2022-05-05 03:18:05 | | | | |
| 3 | Jessie Wu | Meta information modification | 3741 | 2022-05-05 08:10:56 | | | | |
| 4 | Jessie Wu | Meta information modification | 3741 | 2022-05-07 09:50:26 | | |
Video Upload Options
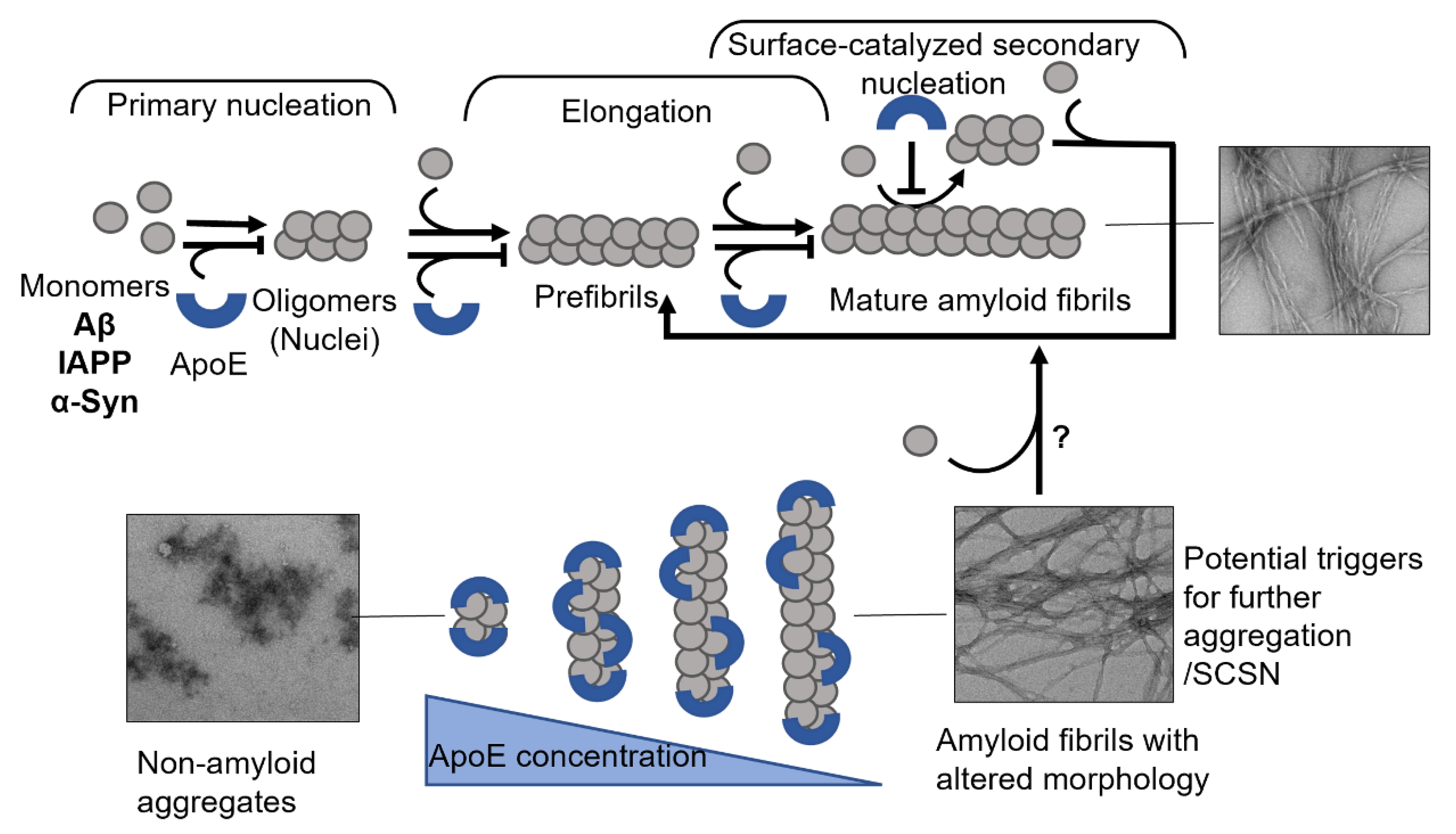
Transthyretin (TTR) is a 55 kDa homotetrameric protein circulating in the plasma and CSF. Its main physiological function is to transport thyroid hormone thyroxine (T4) and retinol-binding protein. Apolipoprotein E (ApoE) is a 34 kDa lipid-binding and transport protein found in both serum and CSF. Amyloid formation is a pathological process associated with a wide range of degenerative disorders, including Alzheimer’s disease, Parkinson’s disease, and diabetes mellitus type 2. During disease progression, abnormal accumulation and deposition of proteinaceous material are accompanied by tissue degradation, inflammation, and dysfunction. Agents that can interfere with the process of amyloid formation or target already formed amyloid assemblies are consequently of therapeutic interest. TTR and ApoE are among the few endogenous proteins with anti-amyloidogenic activity, therefore understanding their role and mechanism of action on amyloid formation can help in developing new therapeutic strategies.
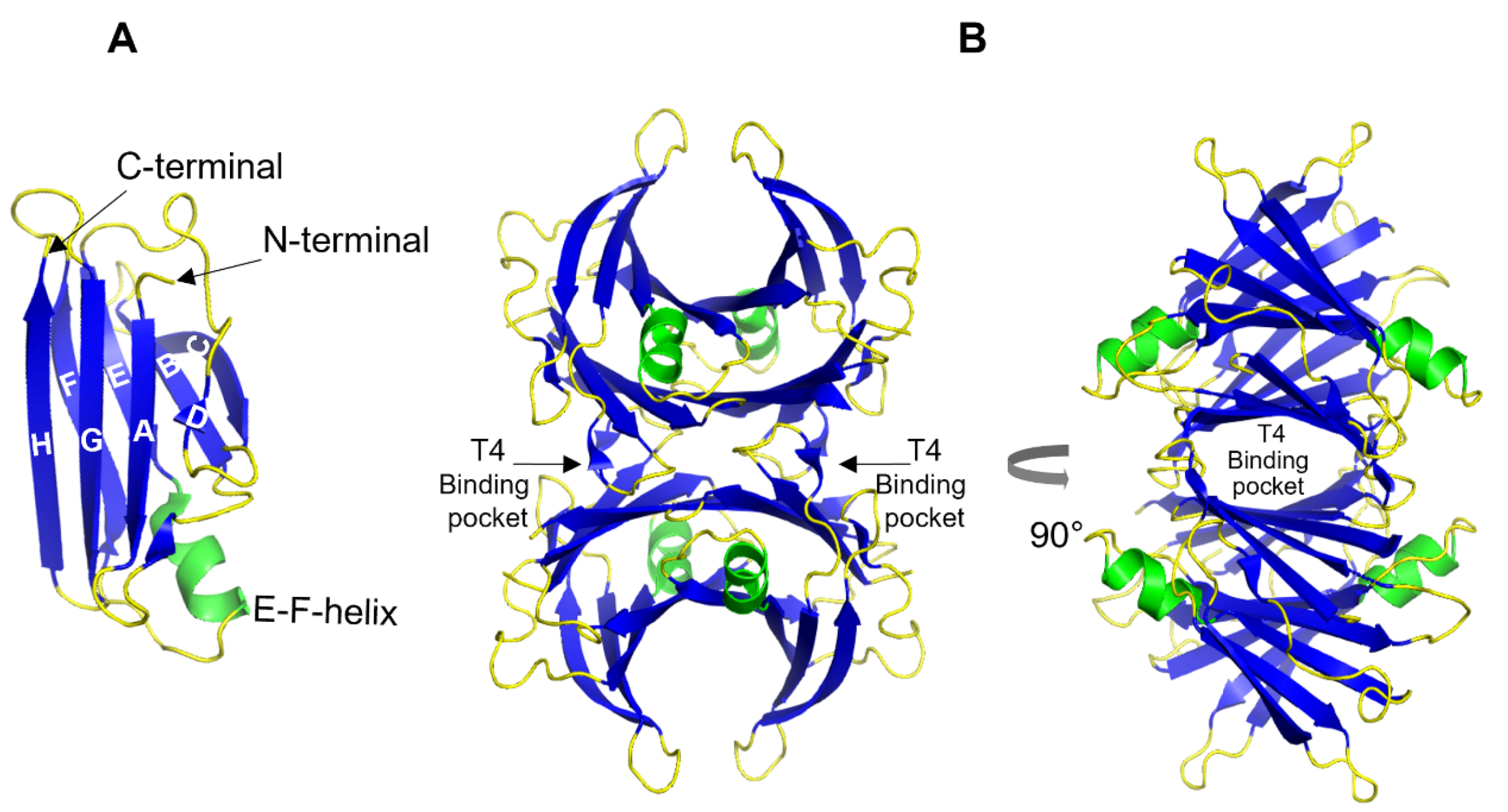
1. Transthyretin
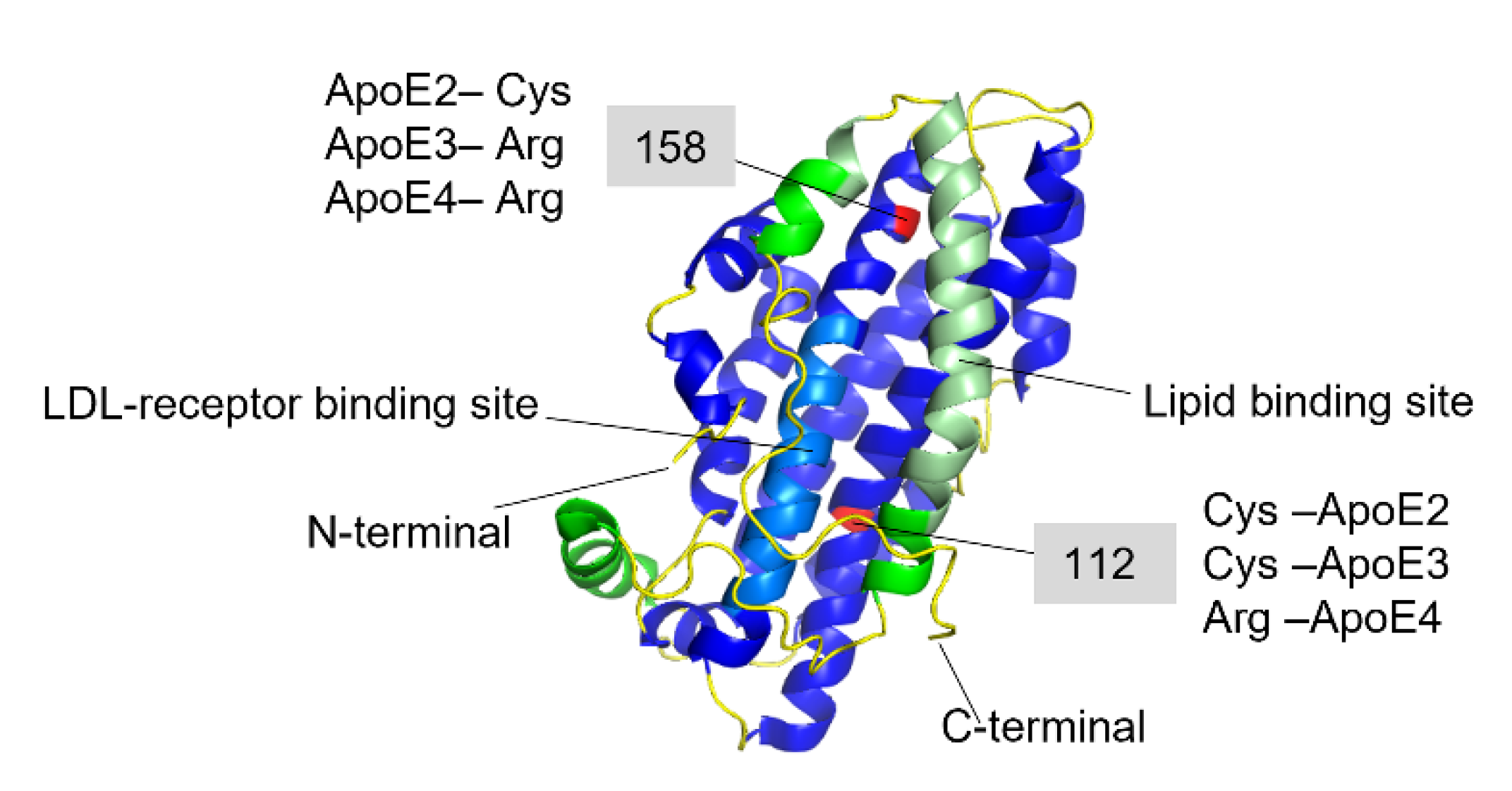
2. Apolipoprotein E
References
- Aleshire, S.L.; Bradley, C.A.; Richardson, L.D.; Parl, F.F. Localization of human prealbumin in choroid plexus epithelium. J. Histochem. Cytochem. Off. J. Histochem. Soc. 1983, 31, 608–612.
- Dickson, P.W.; Schreiber, G. High levels of messenger RNA for transthyretin (prealbumin) in human choroid plexus. Neurosci. Lett. 1986, 66, 311–315.
- Westermark, G.T.; Westermark, P. Transthyretin and Amyloid in the Islets of Langerhans in Type-2 Diabetes. Exp. Diabetes Res. 2008, 2008, 429274.
- Dwork, A.J.; Cavallaro, T.; Martone, R.L.; Goodman, D.S.; Schon, E.A.; Herbert, J. Distribution of transthyretin in the rat eye. Investig. Ophthalmol. Vis. Sci. 1990, 31, 489–496.
- Murakami, T.; Ohsawa, Y.; Zhenghua, L.; Yamamura, K.-I.; Sunada, Y. The transthyretin gene is expressed in Schwann cells of peripheral nerves. Brain Res. 2010, 1348, 222–225.
- Li, X.; Masliah, E.; Reixach, N.; Buxbaum, J.N. Neuronal Production of Transthyretin in Human and Murine Alzheimer’s Disease: Is It Protective? J. Neurosci. 2011, 31, 12483–12490.
- Blake, C.C.F.; Geisow, M.J.; Oatley, S.J.; Rérat, B.; Rérat, C. Structure of prealbumin: Secondary, tertiary and quaternary interactions determined by Fourier refinement at 1.8 Å. J. Mol. Biol. 1978, 121, 339–356.
- Blake, C.C.F.; Swan, I.D.A.; Rerat, C.; Berthou, J.; Laurent, A.; Rerat, B. An X-ray study of the subunit structure of prealbumin. J. Mol. Biol. 1971, 61, 217–224.
- Yee, A.W.; Aldeghi, M.; Blakeley, M.P.; Ostermann, A.; Mas, P.J.; Moulin, M.; De Sanctis, D.; Bowler, M.W.; Mueller-Dieckmann, C.; Mitchell, E.P.; et al. A molecular mechanism for transthyretin amyloidogenesis. Nat. Commun. 2019, 10, 925.
- Westermark, P.; Sletten, K.; Johansson, B.; Cornwell, G.G. Fibril in senile systemic amyloidosis is derived from normal transthyretin. Proc. Natl. Acad. Sci. USA 1990, 87, 2843–2845.
- Ando, Y.; Nakamura, M.; Araki, S. Transthyretin-Related Familial Amyloidotic Polyneuropathy. Arch. Neurol. 2005, 62, 1057–1062.
- Skinner, M. Familial amyloidotic cardiomyopathy. J. Lab. Clin. Med. 1991, 117, 171–172.
- Quintas, A.; Saraiva, M.J.; Brito, R.M.M. The Tetrameric Protein Transthyretin Dissociates to a Non-native Monomer in Solution: A novel model for amyloidogenesis. J. Biol. Chem. 1999, 274, 32943–32949.
- Colon, W.; Kelly, J.W. Partial denaturation of transthyretin is sufficient for amyloid fibril formation in vitro. Biochemistry 1992, 31, 8654–8660.
- Lai, Z.; Colón, W.; Kelly, J.W. The Acid-Mediated Denaturation Pathway of Transthyretin Yields a Conformational Intermediate That Can Self-Assemble into Amyloid. Biochemistry 1996, 35, 6470–6482.
- Liu, K.; Cho, H.S.; Lashuel, H.A.; Kelly, J.W.; Wemmer, D.E. A glimpse of a possible amyloidogenic intermediate of transthyretin. Nat. Struct. Biol. 2000, 7, 754–757.
- Jiang, X.; Smith, C.S.; Petrassi, H.M.; Hammarström, P.; White, J.T.; Sacchettini, J.C.; Kelly, J.W. An Engineered Transthyretin Monomer that Is Nonamyloidogenic, Unless It Is Partially Denatured. Biochemistry 2001, 40, 11442–11452.
- Buxbaum, J.N.; Johansson, J. Transthyretin and BRICHOS: The Paradox of Amyloidogenic Proteins with Anti-Amyloidogenic Activity for Aβ in the Central Nervous System. Front. Neurosci. 2017, 11, 119.
- Costa, R.; Gonçalves, A.; Saraiva, M.; Cardoso, I. Transthyretin binding to A-Beta peptide—Impact on A-Beta fibrillogenesis and toxicity. FEBS Lett. 2008, 582, 936–942.
- Du, J.; Cho, P.Y.; Yang, D.T.; Murphy, R.M. Identification of beta-amyloid-binding sites on transthyretin. Protein Eng. Des. Sel. 2012, 25, 337–345.
- Du, J.; Murphy, R.M. Characterization of the Interaction of β-Amyloid with Transthyretin Monomers and Tetramers. Biochemistry 2010, 49, 8276–8289.
- Ghadami, S.A.; Chia, S.; Ruggeri, F.S.; Meisl, G.; Bemporad, F.; Habchi, J.; Cascella, R.; Dobson, C.M.; Vendruscolo, M.; Knowles, T.P.J.; et al. Transthyretin Inhibits Primary and Secondary Nucleations of Amyloid-β Peptide Aggregation and Reduces the Toxicity of Its Oligomers. Biomacromolecules 2020, 21, 1112–1125.
- Li, X.; Buxbaum, J.N. Transthyretin and the brain re-visited: Is neuronal synthesis of transthyretin protective in Alzheimer’s disease? Mol. Neurodegener. 2011, 6, 79.
- Schwarzman, A.L.; Gregori, L.; Vitek, M.P.; Lyubski, S.; Strittmatter, W.J.; Enghilde, J.J.; Bhasin, R.; Silverman, J.; Weisgraber, K.H.; Coyle, P.K.; et al. Transthyretin sequesters amyloid beta protein and prevents amyloid formation. Proc. Natl. Acad. Sci. USA 1994, 91, 8368–8372.
- Gião, T.; Saavedra, J.; Cotrina, E.; Quintana, J.; Llop, J.; Arsequell, G.; Cardoso, I. Undiscovered Roles for Transthyretin: From a Transporter Protein to a New Therapeutic Target for Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 2075.
- Gloeckner, S.F.; Meyne, F.; Wagner, F.; Heinemann, U.; Krasnianski, A.; Meissner, B.; Zerr, I. Quantitative analysis of transthyretin, tau and amyloid-beta in patients with dementia. J. Alzheimer’s Dis. 2008, 14, 17–25.
- Merched, A.; Serot, J.M.; Visvikis, S.; Aguillon, D.; Faure, G.; Siest, G. Apolipoprotein E, transthyretin and actin in the CSF of Alzheimer’s patients: Relation with the senile plaques and cytoskeleton biochemistry. FEBS Lett. 1998, 425, 225–228.
- Palha, J.; Moreira, P.; Wisniewski, T.; Frangione, B.; Saraiva, M.J. Transthyretin gene in Alzheimer’s disease patients. Neurosci. Lett. 1996, 204, 212–214.
- Buxbaum, J.N.; Ye, Z.; Reixach, N.; Friske, L.; Levy, C.; Das, P.; Golde, T.; Masliah, E.; Roberts, A.R.; Bartfai, T. Transthyretin protects Alzheimer’s mice from the behavioral and biochemical effects of Aβ toxicity. Proc. Natl. Acad. Sci. USA 2008, 105, 2681–2686.
- Stein, T.; Anders, N.J.; DeCarli, C.; Chan, S.L.; Mattson, M.P.; Johnson, J.A. Neutralization of Transthyretin Reverses the Neuroprotective Effects of Secreted Amyloid Precursor Protein (APP) in APPSw Mice Resulting in Tau Phosphorylation and Loss of Hippocampal Neurons: Support for the Amyloid Hypothesis. J. Neurosci. 2004, 24, 7707–7717.
- Choi, S.H.; Leight, S.N.; Lee, V.M.-Y.; Li, T.; Wong, P.C.; Johnson, J.A.; Saraiva, M.J.; Sisodia, S.S. Accelerated Aβ Deposition in APPswe/PS1ΔE9 Mice with Hemizygous Deletions of TTR (Transthyretin). J. Neurosci. 2007, 27, 7006–7010.
- Kerridge, C.; Belyaev, N.D.; Nalivaeva, N.N.; Turner, A.J. The Aβ-clearance protein transthyretin, like neprilysin, is epigenetically regulated by the amyloid precursor protein intracellular domain. J. Neurochem. 2014, 130, 419–431.
- Cascella, R.; Conti, S.; Mannini, B.; Li, X.; Buxbaum, J.N.; Tiribilli, B.; Chiti, F.; Cecchi, C. Transthyretin suppresses the toxicity of oligomers formed by misfolded proteins in vitro. Biochim. Biophys. Acta 2013, 1832, 2302–2314.
- Mazur-Kolecka, B.; Frackowiak, J.; Wiśniewski, H.M. Apolipoproteins E3 and E4 induce, and transthyretin prevents accumulation of the Alzheimer’s β-amyloid peptide in cultured vascular smooth muscle cells. Brain Res. 1995, 698, 217–222.
- Giunta, S.; Valli, M.; Galeazzi, R.; Fattoretti, P.; Corder, E.H.; Galeazzi, L. Transthyretin inhibition of amyloid beta aggregation and toxicity. Clin. Biochem. 2005, 38, 1112–1119.
- Cao, Q.; Anderson, D.H.; Liang, W.Y.; Chou, J.A.; Saelices, L. The inhibition of cellular toxicity of amyloid-β by dissociated transthyretin. J. Biol. Chem. 2020, 295, 14015–14024.
- Mangrolia, P.; Yang, D.T.; Murphy, R.M. Transthyretin variants with improved inhibition of β-amyloid aggregation. Protein Eng. Des. Sel. 2016, 29, 209–218.
- Li, X.; Zhang, X.; Ladiwala, A.R.A.; Du, D.; Yadav, J.K.; Tessier, P.; Wright, P.; Kelly, J.W.; Buxbaum, J.N. Mechanisms of Transthyretin Inhibition of β-Amyloid Aggregation In Vitro. J. Neurosci. 2013, 33, 19423–19433.
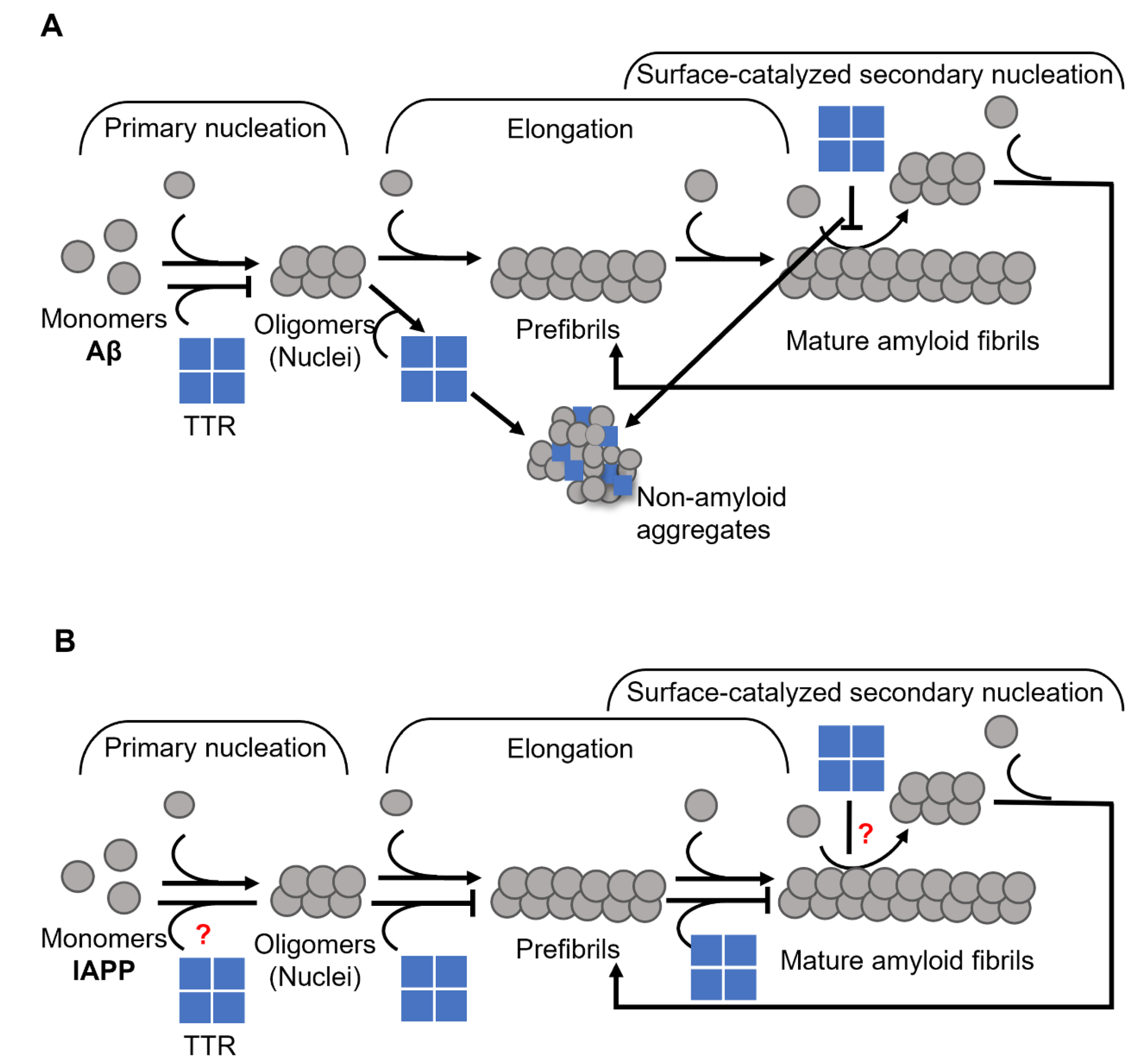
- Nilsson, L.; Pamren, A.; Islam, T.; Brannstrom, K.; Golchin, S.A.; Pettersson, N.; Iakovleva, I.; Sandblad, L.; Gharibyan, A.L.; Olofsson, A. Transthyretin Interferes with Abeta Amyloid Formation by Redirecting Oligomeric Nuclei into Non-Amyloid Aggregates. J. Mol. Biol. 2018, 430, 2722–2733.
- Garai, K.; Posey, A.E.; Li, X.; Buxbaum, J.N.; Pappu, R.V. Inhibition of amyloid beta fibril formation by monomeric human transthyretin. Protein Sci. 2018, 27, 1252–1261.
- Yang, D.T.; Joshi, G.; Cho, P.Y.; Johnson, J.A.; Murphy, R.M. Transthyretin as both a Sensor and a Scavenger of β-Amyloid Oligomers. Biochemistry 2013, 52, 2849–2861.
- Schwarzman, A.L.; Tsiper, M.; Wente, H.; Wang, A.; Vitek, M.P.; Vasiliev, V.; Goldgaber, D. Amyloidogenic and anti-amyloidogenic properties of recombinant transthyretin variants. Amyloid 2004, 11, 1–9.
- Alemi, M.; Silva, S.C.; Santana, I.; Cardoso, I. Transthyretin stability is critical in assisting beta amyloid clearance- Relevance of transthyretin stabilization in Alzheimer’s disease. CNS Neurosci. Ther. 2017, 23, 605–619.
- Rejc, L.; Gómez-Vallejo, V.; Rios, X.; Cossío, U.; Baz, Z.; Mujica, E.; Gião, T.; Cotrina, E.Y.; Jiménez-Barbero, J.; Quintana, J.; et al. Oral Treatment with Iododiflunisal Delays Hippocampal Amyloid-β Formation in a Transgenic Mouse Model of Alzheimer’s Disease: A Longitudinal in vivo Molecular Imaging Study1. J. Alzheimer’s Dis. 2020, 77, 99–112.
- Ribeiro, C.A.; Oliveira, S.M.; Guido, L.F.; Magalhães, A.; Valencia, G.; Arsequell, G.; Saraiva, M.J.; Cardoso, I. Transthyretin Stabilization by Iododiflunisal Promotes Amyloid-β Peptide Clearance, Decreases its Deposition, and Ameliorates Cognitive Deficits in an Alzheimer’s Disease Mouse Model. J. Alzheimer’s Dis. 2014, 39, 357–370.
- Ribeiro, C.A.; Santana, I.; Oliveira, C.; Baldeiras, I.; Moreira, J.; Saraiva, M.J.; Cardoso, I. Transthyretin Decrease in Plasma of MCI and AD Patients: Investigation of Mechanisms for Disease Modulation. Curr. Alzheimer Res. 2012, 9, 881–889.
- Saponaro, F.; Kim, J.H.; Chiellini, G. Transthyretin Stabilization: An Emerging Strategy for the Treatment of Alzheimer’s Disease? Int. J. Mol. Sci. 2020, 21, 8672.
- Wasana Jayaweera, S.; Surano, S.; Pettersson, N.; Oskarsson, E.; Lettius, L.; Gharibyan, A.L.; Anan, I.; Olofsson, A. Mechanisms of Transthyretin Inhibition of IAPP AmyloidFormation. Biomolecules 2021, 11, 411.
- Pate, K.M.; Kim, B.J.; Shusta, E.V.; Murphy, R.M. Transthyretin Mimetics as Anti-β-Amyloid Agents: A Comparison of Peptide and Protein Approaches. ChemMedChem 2018, 13, 968–979.
- Westermark, P.; Wernstedt, C.; Wilander, E.; Hayden, D.W.; O’Brien, T.; Johnson, K.H. Amyloid fibrils in human insulinoma and islets of Langerhans of the diabetic cat are derived from a neuropeptide-like protein also present in normal islet cells. Proc. Natl. Acad. Sci. USA 1987, 84, 3881–3885.
- Westermark, P.; Andersson, A.; Westermark, G.T. Islet Amyloid Polypeptide, Islet Amyloid, and Diabetes Mellitus. Physiol. Rev. 2011, 91, 795–826.
- Westermark, P.; Grimelius, L. The pancreatic islet cells in insular amyloidosis in human diabetic and non-diabetic adults. Acta Pathol. Microbiol. Scand. Sect. A Pathol. 1973, 81, 291–300.
- Jacobsson, B.; Collins, V.P.; Grimelius, L.; Pettersson, T.; Sandstedt, B.; Carlström, A. Transthyretin immunoreactivity in human and porcine liver, choroid plexus, and pancreatic islets. J. Histochem. Cytochem. 1989, 37, 31–37.
- Jain, N.; Ådén, J.; Nagamatsu, K.; Evans, M.L.; Li, X.; McMichael, B.; Ivanova, M.I.; Almqvist, F.; Buxbaum, J.N.; Chapman, M.R. Inhibition of curli assembly and Escherichia coli biofilm formation by the human systemic amyloid precursor transthyretin. Proc. Natl. Acad. Sci. USA 2017, 114, 12184–12189.
- Adams, D.; Polydefkis, M.; González-Duarte, A.; Wixner, J.; Kristen, A.V.; Schmidt, H.H.; Berk, J.L.; López, I.A.L.; Dispenzieri, A.; Quan, D.; et al. Long-term safety and efficacy of patisiran for hereditary transthyretin-mediated amyloidosis with polyneuropathy: 12-month results of an open-label extension study. Lancet Neurol. 2021, 20, 49–59.
- Huang, Y.; Mahley, R.W. Apolipoprotein E: Structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol. Dis. 2014, 72 Pt A, 3–12.
- Fernandez, C.G.; Hamby, M.E.; McReynolds, M.L.; Ray, W.J. The Role of APOE4 in Disrupting the Homeostatic Functions of Astrocytes and Microglia in Aging and Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 14.
- Chen, Y.; Strickland, M.R.; Soranno, A.; Holtzman, D.M. Apolipoprotein E: Structural Insights and Links to Alzheimer Disease Pathogenesis. Neuron 2021, 109, 205–221.
- Weisgraber, K.H. Apolipoprotein E: Structure-Function Relationships. Adv. Protein Chem. 1994, 45, 249–302.
- Huang, Y.; Weisgraber, K.H.; Mucke, L.; Mahley, R.W. Apolipoprotein E: Diversity of Cellular Origins, Structural and Biophysical Properties, and Effects in Alzheimer’s Disease. J. Mol. Neurosci. 2004, 23, 189–204.
- Arendt, T.; Schindler, C.; Brückner, M.K.; Eschrich, K.; Bigl, V.; Zedlick, D.; Marcova, L. Plastic Neuronal Remodeling Is Impaired in Patients with Alzheimer’s Disease Carrying Apolipoprotein ε4 Allele. J. Neurosci. 1997, 17, 516–529.
- Yu, J.-T.; Tan, L.; Hardy, J. Apolipoprotein E in Alzheimer’s Disease: An Update. Annu. Rev. Neurosci. 2014, 37, 79–100.
- Mahley, R.W. Apolipoprotein E: From cardiovascular disease to neurodegenerative disorders. J. Mol. Med. 2016, 94, 739–746.
- Wisniewski, T.; Frangione, B. Apolipoprotein E: A pathological chaperone protein in patients with cerebral and systemic amyloid. Neurosci. Lett. 1992, 135, 235–238.
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923.
- Jones, P.B.; Adams, K.W.; Rozkalne, A.; Spires-Jones, T.L.; Hshieh, T.T.; Hashimoto, T.; von Armin, C.A.; Mielke, M.; Bacskai, B.J.; Hyman, B.T. Apolipoprotein E: Isoform specific differences in tertiary structure and interaction with amyloid-beta in human Alzheimer brain. PLoS ONE 2011, 6, e14586.
- Bales, K.R.; Liu, F.; Wu, S.; Lin, S.; Koger, D.; DeLong, C.; Hansen, J.C.; Sullivan, P.M.; Paul, S.M. Human APOE isoform-dependent effects on brain beta-amyloid levels in PDAPP transgenic mice. J. Neurosci. 2009, 29, 6771–6779.
- Castellano, J.M.; Kim, J.; Stewart, F.R.; Jiang, H.; DeMattos, R.B.; Patterson, B.W.; Fagan, A.M.; Morris, J.C.; Mawuenyega, K.G.; Cruchaga, C.; et al. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci. Transl. Med. 2011, 3, 89ra57.
- Hashimoto, T.; Serrano-Pozo, A.; Hori, Y.; Adams, K.W.; Takeda, S.; Banerji, A.O.; Mitani, A.; Joyner, D.; Thyssen, D.H.; Bacskai, B.J.; et al. Apolipoprotein E, Especially Apolipoprotein E4, Increases the Oligomerization of Amyloid β Peptide. J. Neurosci. 2012, 32, 15181–15192.
- Ghosh, S.; Sil, T.B.; Dolai, S.; Garai, K. High-affinity multivalent interactions between apolipoprotein E and the oligomers of amyloid-beta. FEBS J. 2019, 286, 4737–4753.
- Garai, K.; Verghese, P.B.; Baban, B.; Holtzman, D.M.; Frieden, C. The Binding of Apolipoprotein E to Oligomers and Fibrils of Amyloid-β Alters the Kinetics of Amyloid Aggregation. Biochemistry 2014, 53, 6323–6331.
- Lee, C.Y.; Tse, W.; Smith, J.D.; Landreth, G.E. Apolipoprotein E promotes beta-amyloid trafficking and degradation by modulating microglial cholesterol levels. J. Biol. Chem. 2012, 287, 2032–2044.
- Bruinsma, I.B.; Wilhelmus, M.M.; Kox, M.; Veerhuis, R.; de Waal, R.M.; Verbeek, M.M. Apolipoprotein E protects cultured pericytes and astrocytes from D-Abeta(1-40)-mediated cell death. Brain Res. 2010, 1315, 169–180.
- Ly, S.; Altman, R.; Petrlova, J.; Lin, Y.; Hilt, S.; Huser, T.; Laurence, T.A.; Voss, J.C. Binding of apolipoprotein E inhibits the oligomer growth of amyloid-beta peptide in solution as determined by fluorescence cross-correlation spectroscopy. J. Biol. Chem. 2013, 288, 11628–11635.
- Liao, F.; Yoon, H.; Kim, J. Apolipoprotein E metabolism and functions in brain and its role in Alzheimer’s disease. Curr. Opin. Lipidol. 2017, 28, 60–67.
- Ma, Q.; Zhao, Z.; Sagare, A.P.; Wu, Y.; Wang, M.; Owens, N.C.; Verghese, P.B.; Herz, J.; Holtzman, D.M.; Zlokovic, B.V. Blood-brain barrier-associated pericytes internalize and clear aggregated amyloid-beta42 by LRP1-dependent apolipoprotein E isoform-specific mechanism. Mol. Neurodegener. 2018, 13, 57.
- Potter, H.; Ma, J.; Das, S.; Kayyali, U. The involvement of amyloid associated proteins in the formation of beta-protein filaments in Alzheimer’s disease. Prog. Clin. Biol. Res. 1994, 390, 57–71.
- Holtzman, D.M.; Fagan, A.M.; Mackey, B.; Tenkova, T.; Sartorius, L.; Paul, S.M.; Bales, K.; Ashe, K.H.; Irizarry, M.C.; Hyman, B.T. Apolipoprotein E facilitates neuritic and cerebrovascular plaque formation in an Alzheimer’s disease model. Ann. Neurol. 2000, 47, 739–747.
- Rall, S.C., Jr.; Weisgraber, K.H.; Mahley, R.W. Human apolipoprotein E. The complete amino acid sequence. J. Biol. Chem. 1982, 257, 4171–4178.
- Weisgraber, K.H.; Rall, S.C., Jr.; Mahley, R.W. Human E apoprotein heterogeneity. Cysteine-arginine interchanges in the amino acid sequence of the apo-E isoforms. J. Biol. Chem. 1981, 256, 9077–9083.
- Raulin, A.-C.; Kraft, L.; Al-Hilaly, Y.; Xue, W.-F.; McGeehan, J.; Atack, J.R.; Serpell, L. The Molecular Basis for Apolipoprotein E4 as the Major Risk Factor for Late-Onset Alzheimer’s Disease. J. Mol. Biol. 2019, 431, 2248–2265.
- Frieden, C.; Garai, K. Concerning the structure of apoE. Protein Sci. 2013, 22, 1820–1825.
- Saito, H.; Dhanasekaran, P.; Baldwin, F.; Weisgraber, K.H.; Phillips, M.C.; Lund-Katz, S. Effects of Polymorphism on the Lipid Interaction of Human Apolipoprotein E. J. Biol. Chem. 2003, 278, 40723–40729.
- Chen, J.; Li, Q.; Wang, J. Topology of human apolipoprotein E3 uniquely regulates its diverse biological functions. Proc. Natl. Acad. Sci. USA 2011, 108, 14813–14818.
- Henry, N.; Krammer, E.-M.; Stengel, F.; Adams, Q.; Van Liefferinge, F.; Hubin, E.; Chaves, R.; Efremov, R.; Aebersold, R.; Vandenbussche, G.; et al. Lipidated apolipoprotein E4 structure and its receptor binding mechanism determined by a combined cross-linking coupled to mass spectrometry and molecular dynamics approach. PLoS Comput. Biol. 2018, 14, e1006165.
- Weisgraber, K.H. Apolipoprotein E distribution among human plasma lipoproteins: Role of the cysteine-arginine interchange at residue 112. J. Lipid Res. 1990, 31, 1503–1511.
- Dong, L.-M.; Parkin, S.; Trakhanov, S.D.; Rupp, B.; Simmons, T.; Arnold, K.S.; Newhouse, Y.M.; Innerarity, T.L.; Weisgraber, K.H. Novel mechanism for defective receptor binding of apolipoprotein E2 in type III hyperlipoproteinemia. Nat. Struct. Mol. Biol. 1996, 3, 718–722.
- Dong, L.-M.; Weisgraber, K.H. Human Apolipoprotein E4 Domain Interaction: Arginine 61 and glutamic acid 255 interact to direct the preference for very low density lipoproteins. J. Biol. Chem. 1996, 271, 19053–19057.
- Raussens, V.; Drury, J.; Forte, T.M.; Choy, N.; Goormaghtigh, E.; Ruysschaert, J.-M.; Narayanaswami, V. Orientation and mode of lipid-binding interaction of human apolipoprotein E C-terminal domain. Biochem. J. 2005, 387, 747–754.
- Narayanaswami, V.; Maiorano, J.N.; Dhanasekaran, P.; Ryan, R.O.; Phillips, M.C.; Lund-Katz, S.; Davidson, W. Helix Orientation of the Functional Domains in Apolipoprotein E in Discoidal High Density Lipoprotein Particles. J. Biol. Chem. 2004, 279, 14273–14279.
- Hatters, D.M.; Voss, J.C.; Budamagunta, M.S.; Newhouse, Y.N.; Weisgraber, K.H. Insight on the Molecular Envelope of Lipid-Bound Apolipoprotein E from Electron Paramagnetic Resonance Spectroscopy. J. Mol. Biol. 2009, 386, 261–271.
- Frieden, C.; Wang, H.; Ho, C.M.W. A mechanism for lipid binding to apoE and the role of intrinsically disordered regions coupled to domain–domain interactions. Proc. Natl. Acad. Sci. USA 2017, 114, 6292–6297.
- Peters-Libeu, C.A.; Newhouse, Y.; Hatters, D.M.; Weisgraber, K.H. Model of Biologically Active Apolipoprotein E Bound to Dipalmitoylphosphatidylcholine. J. Biol. Chem. 2006, 281, 1073–1079.
- Clément-Collin, V.; Barbier, A.; Dergunov, A.D.; Visvikis, A.; Siest, G.; Desmadril, M.; Takahashi, M.; Aggerbeck, L.P. The structure of human apolipoprotein E2, E3 and E4 in solution. Multidomain organization correlates with the stability of apoE structure. Biophys. Chem. 2006, 119, 170–185.
- Hatters, D.M.; Zhong, N.; Rutenber, E.; Weisgraber, K.H. Amino-terminal Domain Stability Mediates Apolipoprotein E Aggregation into Neurotoxic Fibrils. J. Mol. Biol. 2006, 361, 932–944.
- Islam, T.; Gharibyan, A.L.; Golchin, S.A.; Pettersson, N.; Brännström, K.; Hedberg, I.; Virta, M.; Olofsson, L.; Olofsson, A. Apolipoprotein E impairs amyloid-β fibril elongation and maturation. FEBS J. 2020, 287, 1208–1219.
- Holtzman, D.M.; Bales, K.R.; Tenkova, T.; Fagan, A.M.; Parsadanian, M.; Sartorius, L.J.; Mackey, B.; Olney, J.; McKeel, D.; Wozniak, D.; et al. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 2892–2897.
- Liao, F.; Li, A.; Xiong, M.; Bien-Ly, N.; Jiang, H.; Zhang, Y.; Finn, M.B.; Hoyle, R.; Keyser, J.; Lefton, K.B.; et al. Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. J. Clin. Investig. 2018, 128, 2144–2155.
- Jiang, Q.; Lee, C.Y.D.; Mandrekar, S.; Wilkinson, B.; Cramer, P.; Zelcer, N.; Mann, K.; Lamb, B.; Willson, T.M.; Collins, J.L.; et al. ApoE Promotes the Proteolytic Degradation of Aβ. Neuron 2008, 58, 681–693.
- Robert, J.; Button, E.B.; Yuen, B.; Gilmour, M.; Kang, K.; Bahrabadi, A.; Stukas, S.; Zhao, W.; Kulic, I.; Wellington, C.L. Clearance of beta-amyloid is facilitated by apolipoprotein E and circulating high-density lipoproteins in bioengineered human vessels. eLife 2017, 6, e29595.
- Wilhelmus, M.M.M.; Otte-Höller, I.; Davis, J.; Van Nostrand, W.E.; De Waal, R.M.W.; Verbeek, M.M. Apolipoprotein E Genotype Regulates Amyloid-β Cytotoxicity. J. Neurosci. 2005, 25, 3621–3627.
- Rasmussen, K.L. Plasma levels of apolipoprotein E, APOE genotype and risk of dementia and ischemic heart disease: A review. Atherosclerosis 2016, 255, 145–155.
- Rasmussen, K.L.; Tybjaerg-Hansen, A.; Nordestgaard, B.G.; Frikke-Schmidt, R. Plasma apolipoprotein E levels and risk of dementia: A Mendelian randomization study of 106,562 individuals. Alzheimers Dement. 2018, 14, 71–80.
- Gupta, V.B.; Wilson, A.C.; Burnham, S.; Hone, E.; Pedrini, S.; Laws, S.M.; Lim, W.L.F.; Rembach, A.; Rainey-Smith, S.; Ames, D.; et al. Follow-up plasma apolipoprotein E levels in the Australian Imaging, Biomarkers and Lifestyle Flagship Study of Ageing (AIBL) cohort. Alzheimer’s Res. Ther. 2015, 7, 16.
- Wolters, F.J.; Koudstaal, P.J.; Hofman, A.; Van Duijn, C.M.; Ikram, M.A. Serum apolipoprotein E is associated with long-term risk of Alzheimer’s disease: The Rotterdam Study. Neurosci. Lett. 2016, 617, 139–142.
- Martínez-Morillo, E.; Hansson, O.; Atagi, Y.; Bu, G.; Minthon, L.; Diamandis, E.P.; Nielsen, H.M. Total apolipoprotein E levels and specific isoform composition in cerebrospinal fluid and plasma from Alzheimer’s disease patients and controls. Acta Neuropathol. 2014, 127, 633–643.
- Bertrand, P.; Poirier, J.; Oda, T.; Finch, C.E.; Pasinetti, G. Association of apolipoprotein E genotype with brain levels of apolipoprotein E and apolipoprotein J (clusterin) in Alzheimer disease. Brain Res. Mol. Brain Res. 1995, 33, 174–178.
- Talwar, P.; Sinha, J.; Grover, S.; Agarwal, R.; Kushwaha, S.; Srivastava, M.V.P.; Kukreti, R. Meta-analysis of apolipoprotein E levels in the cerebrospinal fluid of patients with Alzheimer’s disease. J. Neurol. Sci. 2016, 360, 179–187.
- Cruchaga, C.; Kauwe, J.S.; Nowotny, P.; Bales, K.; Pickering, E.H.; Mayo, K.; Bertelsen, S.; Hinrichs, A.; Fagan, A.M.; Holtzman, D.M.; et al. Cerebrospinal fluid APOE levels: An endophenotype for genetic studies for Alzheimer’s disease. Hum. Mol. Genet. 2012, 21, 4558–4571.
- Sihlbom, C.; Davidsson, P.; Sjögren, M.; Wahlund, L.-O.; Nilsson, C.L. Structural and Quantitative Comparison of Cerebrospinal Fluid Glycoproteins in Alzheimer’s Disease Patients and Healthy Individuals. Neurochem. Res. 2008, 33, 1332–1340.
- Akram, A.; Schmeidler, J.; Katsel, P.; Hof, P.R.; Haroutunian, V. Association of ApoE and LRP mRNA levels with dementia and AD neuropathology. Neurobiol. Aging 2012, 33, 628.e1–628.e14.
- Baker-Nigh, A.T.; Mawuenyega, K.G.; Bollinger, J.G.; Ovod, V.; Kasten, T.; Franklin, E.E.; Liao, F.; Jiang, H.; Holtzman, D.; Cairns, N.J.; et al. Human Central Nervous System (CNS) ApoE Isoforms Are Increased by Age, Differentially Altered by Amyloidosis, and Relative Amounts Reversed in the CNS Compared with Plasma. J. Biol. Chem. 2016, 291, 27204–27218.
- Schmidt, C.; Becker, H.; Zerr, I. Cerebrospinal Fluid Apolipoprotein E Concentration and Severity of Cognitive Impairment in Patients With Newly Diagnosed Alzheimer’s Disease. Am. J. Alzheimer’s Dis. Other Dement. 2014, 29, 54–60.
- Arboleda-Velasquez, J.F.; Lopera, F.; O’Hare, M.; Delgado-Tirado, S.; Marino, C.; Chmielewska, N.; Saez-Torres, K.L.; Amarnani, D.; Schultz, A.P.; Sperling, R.A.; et al. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: A case report. Nat. Med. 2019, 25, 1680–1683.
- Lei, P.; Wu, W.-H.; Li, R.-W.; Ma, J.-W.; Yu, Y.-P.; Cui, W.; Zhao, Y.-F.; Li, Y.-M. Prevention and promotion effects of apolipoprotein E4 on amylin aggregation. Biochem. Biophys. Res. Commun. 2008, 368, 414–418.
- Emamzadeh, F.N.; Aojula, H.; McHugh, P.; Allsop, D. Effects of different isoforms of apoE on aggregation of the α-synuclein protein implicated in Parkinson’s disease. Neurosci. Lett. 2016, 618, 146–151.
- Gharibyan, A.L.; Islam, T.; Pettersson, N.; Golchin, S.A.; Lundgren, J.; Johansson, G.; Genot, M.; Schultz, N.; Wennström, M.; Olofsson, A. Apolipoprotein E Interferes with IAPP Aggregation and Protects Pericytes from IAPP-Induced Toxicity. Biomolecules 2020, 10, 134.
- Gallardo, G.; Schlüter, O.M.; Südhof, T.C. A molecular pathway of neurodegeneration linking α-synuclein to ApoE and Aβ peptides. Nat. Neurosci. 2008, 11, 301–308.
- Williams, T.; Borchelt, D.R.; Chakrabarty, P. Therapeutic approaches targeting Apolipoprotein E function in Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 8.

