1. Transthyretin
Transthyretin (T
TR) is produced mainly in the liver
[1][49] wherefrom it is secreted into the blood with concentration levels ranging between 4–7 µM. In the CSF, the concentration of TTR is about 20 times lower and it is primarily synthesized in the choroid plexus
[2][50]. TTR is also expressed by the islet of Langerhans of the pancreas
[3][51] as well as within the retina of the eye
[4][52]. TTR gene expression can also be found in Schwann cells, dorsal root ganglia
[5][53], and cortical and hippocampal neurons in response to Aβ-induced stress
[6][54]. The structure of TTR is well studied and was first solved by X-ray crystallography in 1978
[7][55] (
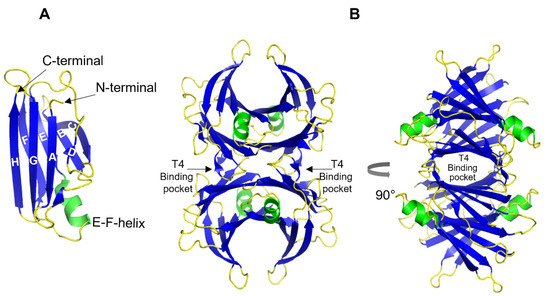
Figure 1). Each TTR monomer consists of 127 amino acid residues forming eight β-strands named A-H and one short α-helix between E and F strands
[7][8][55,56] (
Figure 1A). The β-strands CBEF and DAGH are arranged into two β-sheets. Two monomers form a dimer via hydrogen bonds between antiparallel H-H’ and F-F’ strands. The functional tetramer is formed from two dimers predominantly by hydrophobic interactions between the residues of A-B and G-H loops, producing a central hydrophobic pocket with two binding sites for thyroxin hormone
[7][8][55,56] (
Figure 1B). Both monomer–monomer and dimer–dimer interactions are important for the stability of TTR
[9][57].
Figure 1. 3D ribbon diagram of the native human TTR protein. (A). TTR monomer with eight β-strands A-H, and an α-helix between E and F strands. (B). TTR tetramer in two different orientations and indicated thyroxine-binding pocked. Key features are colored as β-strands in blue and α-helices in green. The figures are created from PDB-ID: 1DVQ.
TTR also possesses intrinsic amyloidogenic features and its aggregation is linked to several amyloid disorders including senile systemic amyloidosis (SSA), familial amyloid cardiomyopathy (FAC), and familial amyloid polyneuropathy (FAP)—a hereditary disease variant caused by one of over 100 known point mutations in the
ttr gene
[10][11][12][58,59,60]. The initial and rate-limiting step in TTR aggregation is the dissociation of the native tetramer into monomers that subsequently undergo conformational changes forming aggregation-prone intermediates
[13][14][15][16][17][61,62,63,64,65].
Paradoxically, being an amyloidogenic protein itself, it was found that TTR has an inhibitory effect on AD-associated Aβ aggregation via direct interaction with the latter
[18][19][20][21][22][23][66,67,68,69,70,71]. This discovery suggested a protective role of TTR against AD, where a possible failure of these properties could serve as a precondition for the disease development
[24][32] (the role of TTR in AD was recently reviewed by Giao et al.
[25][72]). This hypothesis is further supported by clinical data showing a significantly reduced level of TTR in the CSF of AD patients compared to healthy individuals
[26][27][73,74]. Moreover, the severity of the disease was inversely correlated with the concentration of TTR
[27][74]; however, no correlation was found between the variants of TTR and AD
[28][75].
Since then, numerous studies have confirmed the protective effect of TTR in animal models of AD. Particularly, overexpression of human TTR in the widely used AD transgenic mouse model APP23 prevents cognitive impairment
[29][76]. Stein et. al. showed that transgenic Tg2576 mice that overexpress mutant amyloid precursor protein (APP) exhibit upregulation of TTR expression, and chronic infusion of an anti-TTR antibody into the hippocampus of these mice exacerbates Aβ pathology, as the concentration of free TTR is decreased
[30][77]. In addition, hemizygous TTR deletion in APPswe/PS1deltaE9 mice resulted in earlier deposition of Aβ fibrils compared to control mice
[31][78]. It has been shown that the expression level of TTR, similar to Aβ-degrading neprilysin, is controlled by the intracellular domain of APP
[32][79]; thus, overexpression of APP in Tg2576 mice could mediate the up-regulation of TTR
[30][77].
The protective effect of TTR against Aβ-induced cytotoxicity has also been investigated in cellular models
[19][33][34][35][36][35,67,80,81,82]. Together, these findings suggest a functional connection between Aβ and TTR, and their interactions have been studied extensively in vitro
[21][22][37][38][39][40][33,34,37,69,70,83]. The results support an inhibiting effect of TTR on Aβ amyloid formation. However, in this context, there are several controversies in the field. By using nanoparticle tracking, Yang et al., showed that partially aggregated Aβ binds to TTR more efficiently than the monomeric or heavily aggregated form of Aβ, and monomeric TTR binds more Aβ than tetrameric form
[41][84]. The authors suggest a mechanism where the EF-strand helix/loop of TTR tetramer acts as a sensor in the presence of Aβ oligomers, the binding of which triggers destabilization of TTR tetramer and exposure of inner hydrophobic sheets, thus increasing the oligomer binding capacity and arresting their further aggregation
[41][84]. In contrast to this, Li et al., demonstrated that Aβ monomers bind to the ligand-binding pocket of TTR, thus sequestering monomers from the oligomer formation process
[38][34]. At the same time, they also suggest that both tetrameric and monomeric TTR can bind Aβ oligomers, with greater efficiency for TTR monomers in vitro
[38][34]. In support of this, some data suggest an inverse correlation between TTR stability and the inhibitory potential on Aβ aggregation in vitro
[21][69], and that TTR dissociation into monomers is required for the inhibition of Aβ-induced cytotoxicity, while stable tetrameric variants are nonefficient
[36][82]. In contrast, others show that unstable TTR binds poorly to Aβ
[42][85] and that the stability of tetrameric TTR is critical in Aβ clearance by assisting its internalization and degradation in lysosomes
[43][86]. Moreover, oral administration of the TTR stabilizing drug iododiflunisal promotes Aβ clearance, leading to reduced Aβ deposition and improved cognitive functions in AD mouse models
[44][45][87,88]. In this context, it is interesting to note the work by Li et al.
[38][34], where they show that in vitro the kinetically rather unstable TTR-V122I variant is more efficient in inhibiting Aβ aggregation compared to the more stable TTR-T119M and TTRV30M. However, even the most kinetically stable variant can suppress Aβ aggregation at a TTR:Aβ ratio corresponding to 1:5. The authors also suggest that the observed in vitro interactions of TTR monomers with Aβ oligomers might not reflect in vivo situations where the tetrameric form of wild-type TTR is the dominating variant and dissociation of TTR tetramer is not necessary for its inhibitory effect
[38][34]. Given that in vivo the extracellular Aβ concentration is much lower (within high picomolar-low nanomolar range) and TTR concentration is ranging from 0.25–0.5 μM in CSF and 3–5 μM in human serum, this should provide sufficient condition for performing its protective activity against Aβ aggregation. However, studies on AD patients revealed an inability of TTR to bind its natural ligand
[46][89] and a decreased ratio of folded/monomeric TTR in plasma
[43][86], which suggests a deficit in functional TTR molecules. Thus, stabilizing the TTR could represent one of the possibilities in AD therapy which is discussed in a recent review by Saponaro et al.
[47][90].
In a previous study,
rwe
searchers showed that the inhibition of Aβ fibril formation by TTR primarily involves the nucleation stage and results in the formation of ThT- negative non-amyloid aggregates
[39][37]. However,
reswe
archers also noted that TTR does not affect the process of elongation (
Figure 2A). More recently Ghadami et al., reported that TTR can inhibit both primary and secondary nucleation of Aβ aggregation, at the same time confirming
our
esearchers's observation on the inability of TTR to affect the elongation
[22][70]. Interestingly, by modulating the conditions of nucleation as well as the concentration of TTR, essentially a complete conversion into non-amyloid amorphous Aβ assemblies could be acquired
[39][37].
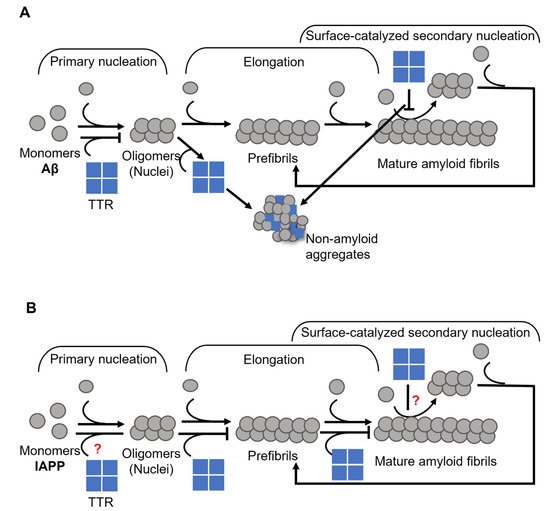
Figure 2. Schematic presentation of suggested mechanisms for TTR interference with (
A) Aβ, where TTR targets both primary and secondary nucleation and re-directs the reaction towards the formation of non-amyloid aggregates (modified from Nilsson et al., 2018
[39][37]), and (
B) IAPP, where TTR specifically targets the elongation process (modified from Wasana Jayaweera et al., 2021
[48][91]) amyloid formation.
Besides the inhibitory effect on Aβ aggregation, there is also evidence that TTR may interfere with other amyloid-forming polypeptides. Recently, Pate et al., showed that both engineered monomeric TTR and cG8 cyclic peptide, which is based on an Aβ-binding domain of TTR, efficiently inhibits amylin aggregation, although both did not affect α-synuclein
[49][92]. Amylin or islet amyloid polypeptide (IAPP) aggregation is associated with type 2 diabetes, where IAPP amyloid affects the islets of Langerhans accompanied by a decreased number of β-cell
[50][51][52][93,94,95]. Normally TTR is expressed by pancreatic α-cells
[53][96], and in much lower levels by β-cells
[3][51]. Interestingly, increased TTR levels were found in β-cell in type-2 diabetes patients’ pancreatic islets with heavy amyloid deposits
[3][51].
In a recently published work,
reswe
archers showed that TTR affects the process of IAPP amyloid formation by targeting elongation
[48][91] (
Figure 2B), which is in stark contrast to its effect on Aβ (see
Figure 2A,B)
[22][39][37,70].
ResWe
archers also showed that the efficacy of TTR displays an inverse correlation with thermodynamic stability, while no such correlation could be noted regarding the rate of dissociation of the tetramer. However, the addition of TTR-stabilizing drugs partly suppresses its efficiency
[48][91] in contrast to the suggested beneficial effect on Aβ
[44][45][87,88], hence supporting that a partial unfolding is required for its inhibitory actions.
These findings suggest that TTR can have an important contribution in regulating the aggregation of certain amyloidogenic polypeptides in vivo. The interaction of TTR with other amyloidogenic proteins, such as bacterial CsgA
[54][97] and HypF-N
[33][35], also indicates its generic nature as an anti-amyloidogenic molecular chaperon.
However, the experimental data available for Aβ and IAPP suggest that although TTR has a common anti-amyloidogenic property on these peptides, the underlying mechanisms of inhibition might be different, and further investigations are needed to gain a better understanding of these mechanisms.
The potential use of TTR as a target to improve its anti-amyloidogenic properties should, however, be handled with care due to its intrinsic amyloidogenic features. Clinical means to improve its anti-amyloidogenic features should consequently also make sure to not enhance its intrinsic amyloidogenic features. The role of TTR as an amyloid-interfering protein may however already be put to the test. The recent development within the techniques of RNA interference has resulted in clinically approved treatments for FAP where the endogenous TTR produced in the liver is suppressed by more than 90%
[55][98]. Here the reverse question is raised where lack of TTR instead potentially may enhance the formation of other amyloids such as Aβ or IAPP.
2. Apolipoprotein E
In the periphery, the majority of circulating ApoE is synthesized by the liver
[56][99], while in the central nervous system (CNS), it is mainly produced by glial cells
[57][100]. Peripheral ApoE plays a critical role in lipid metabolism by transporting lipoproteins into the lymph system and blood and maintaining the cellular uptake of lipoproteins via its primary receptor LDLR (low-density lipoprotein receptor)
[58][59][101,102]. ApoE in the CNS promotes the transport of lipids into neurons, thus participating in neuronal maintenance, synaptic remodeling, repair, and lipid homeostasis in CNS
[60][61][103,104]. Human ApoE exists in three isoforms—ApoE2, ApoE3, and ApoE4, which are encoded by the three alleles ε2, ε3, and ε4, respectively
[62][105]. The dominating isoform in the global population is ApoE3 with an allele frequency of 65–70%, while the ApoE4 variant is found within 15–20% of the population
[63][106]. The discovery of a strong genetic linkage to AD, where the ApoE4 variant was found to be a high genetic risk factor for the development of the disease
[62][64][65][39,105,107], brought this protein into focus. Heterozygous carriers of ε4 allele have about a 2 to 3-fold higher risk of developing AD compared to homozygous ApoE3 carriers, while for homozygous ε4 allele carriers the risk increases about 10-fold compared to non- ε4 carriers
[65][107] and 40–65% of all AD patients are carriers of at least one copy of ApoE4
[66][108]. In contrast, ApoE2 is considered protective
[62][105] and the prevalence of AD development within this group is lower as compared to ApoE3.
ApoE has been found co-localized with Aβ-amyloid plaques with relative prevalence of ApoE4 > ApoE3 > ApoE2
[67][68][109,110]. These results suggested that ApoE might play a specific role in Aβ amyloid formation and related AD pathology, which has been explored in numerous studies
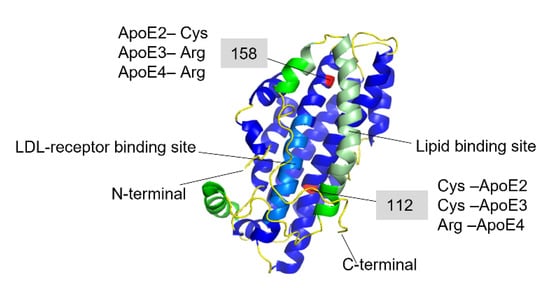
[64][68][69][70][71][72][73][74][75][76][77][78][39,40,41,42,110,111,112,113,114,115,116,117]. The amino acid differences between the three isoforms are only at positions 112 and 158, where ApoE2 has cysteine residues at both positions, ApoE3 has a cysteine at positions 112 and an arginine at position 158, and ApoE4 has arginine residues at both positions (
Figure 3)
[62][79][80][105,118,119].
Figure 3. Full-length structure of Apolipoprotein E3 (PDB-ID: 2L7B). The N-terminal domain is colored in blue where the light-blue region is the LDL-receptor binding site, and the C-terminal domain is colored in green, where the light green region is the lipid-binding site. The red-colored residues at 112 and 158 positions are the varying residues between the three ApoE variants.
These seemingly small differences in the sequence affect the ApoE receptor and lipid binding abilities and functionality
[62][81][82][83][105,120,121,122]. The ApoE molecule consists of two distinct structural N- and C-terminal domains linked together by an unstructured hinge region, which is supporting the mobility of the structure. The N-terminal domain represents a bundle of four α-helixes, comprising positions 1–191, where the LDL-receptor binding region is located (residues 134–150)
[62][84][105,123]. The C-terminal amphipathic α-helical domain consists of residues 210–299 with a largely exposed hydrophobic surface, where the lipid-binding region is located within the residues 244–272
[62][84][105,123]. When ApoE binds to the lipids the whole molecule undergoes conformational changes to adopt a biologically active form, which is necessary for recognition and binding to the LDL-receptors and internalization
[85][124]. Although the isoform-specific residues are located within the N-terminal domain, the preferences for lipid and lipoprotein binding are different between the three ApoE isoforms. ApoE2 and ApoE3 variants are mostly associated with high-density lipoproteins (HDL), while ApoE4 prefers low-density lipoproteins and very-low-density lipoproteins (LDL, VLDL)
[86][125]. These preferences are most likely attributed to differential conformational changes of the whole ApoE molecule provided by the residues at positions 112 and 158. The X-ray crystallography analysis showed that in the dominating isoform-ApoE3 (associated with normal lipid and lipoprotein metabolism), there is a salt bridge formation between the Arg-158 and Asp–154. This salt bridge seems to be critical for LDL-receptor recognizing and binding
[63][87][106,126]. In the ApoE2 variant, Arg to Cys substitution at position 158 leads to disruption of the natural salt bridge and formation of a new bridge between the Asp154-Arg150. This in turn impairs the binding ability of ApoE2 to the LDL-receptor, which is associated with type III hyperlipoproteinemia
[87][126]. In ApoE4, Arg112 causes a rearrangement of the N-terminal Arg61 side chain, which is exposed towards Glu255 in the C-terminal domain leading to a salt bridge formation between these two residues
[88][127]. This interaction between the two domains is responsible for the preferential binding of ApoE4 to VLDL
[83][88][122,127]. In general, ApoE4 is shown to have higher affinity to the lipids and lipoprotein particles, regardless of their size
[83][122]. On the other hand, it has been suggested that the unique interaction between the two domains leads to accelerated catabolism of ApoE4 and consequently, elevated cholesterol and LDL levels in plasma
[83][88][122,127].
Lipid binding to the C-terminal is a requirement for the ApoE molecule to gain LDL-receptor binding ability. When bound to the lipids, the C-terminal α-helices are oriented perpendicularly to the acyl chain of the lipids
[89][90][128,129]. For the N-terminal domain, several models were suggested to adapt for lipid-bound ApoE. Calorimetric studies showed that ApoE gains an extended conformation due to the four-helix bundle opening, which allows wrapping around the lipid bilayer
[90][129]. In contrast to this, other studies suggested a hairpin conformation of ApoE in the lipid-bound state
[91][92][93][130,131,132]. In a more recent study by Henry et al.
[85][124], it was proposed that both the open and compact hairpin conformations may co-exist in a dynamic equilibrium, which is shifted to the opened hairpin model in the presence of the LDL-receptor. Thus, both conformations may be part of a regulation mechanism of ApoE function at the surface of lipids
[85][124].
Further, differences in stability between the ApoE variants indicate that ApoE4 is less stable and that it also may aggregate into amyloid-like fibrils
[94][95][133,134]. In contrast, a recent study revealed no significant differences between the recombinant ApoE variants at the structural or conformational level
[81][120].
It has been shown that ApoE can directly interfere with the Aβ aggregation process by inhibiting or slowing down fibril formation
[64][69][70][71][74][77][96][39,40,41,42,113,116,135]. However, the impact of this interaction on disease progression remains unclear. Several studies suggest that ApoE acts as a pathological chaperone, although to different extents depending on the variant. For example, studies on AD mouse models revealed that all ApoE-carrying AD mice developed amyloid plaques with the highest load observed in ApoE4 animals, while the complete knockout of the ApoE gene significantly reduced plaque formation and other signs of disease
[67][97][109,136]. In agreement with this, administration of ApoE-specific antibodies promoted efficient clearance of Aβ plaques
[98][137]. In contrast, several studies indicate that ApoE plays an important role in the degradation and clearance of Aβ amyloid
[68][72][99][100][110,111,138,139]. For example, it has been shown that ApoE protects human pericytes against Aβ-induced cytotoxicity
[73][112] and maintains a receptor-mediated in-pericyte clearance of Aβ aggregates
[76][115], with significantly weaker effects detected for ApoE4 compared to the other variants
[73][76][112,115]. Interestingly, cells with the ε4 genotype expressed lower levels of ApoE and exhibited a higher vulnerability to Aβ toxicity compared to cells with ε2 or ε3 genotype
[73][101][112,140]. Moreover, a low level of ApoE has been considered as a general risk factor for AD irrespective of isoform
[102][103][141,142]. Therefore, low ApoE expression encoded by the ε4 allele could explain its association to AD pathology as in this case the levels of functional ApoE4 would be insufficient to perform its protective function. However, reports on the levels of ApoE in AD also remain controversial. Several studies link low ApoE levels in plasma to an increased risk of AD and dementia
[104][105][143,144], while others argue that the low levels are specifically associated with the ApoE4 isoform and that there is no difference in ApoE levels in AD vs non-AD individuals
[106][145]. Similarly, conflicting results are reported for CNS ApoE levels in AD, shown to be either decreased
[107][108][109][146,147,148], increased
[110][111][112][149,150,151], or unchanged
[106][113][145,152].
In support of a protective role of ApoE, a recent case report described an individual carrying the ApoE3 Christchurch variant with no signs of AD, despite expectations to develop early-onset AD due to an aggressive familial presenilin mutation
[114][153]. These data further highlight the clinical importance of ApoE both in modulating the amyloid formation and disease progression, as well as implying that ApoE generally has a protective role against amyloid formation and toxicity, with a loss of function for ApoE4.
In vitro, the direct interaction of ApoE with Aβ has an inhibitory effect on the amyloid formation process
[40][70][71][96][41,42,83,135]. Our group recently reported that ApoE targets both the process of Aβ nucleation and fibril elongation, and effectively prevents the maturation of amyloids in a concentration-dependent manner
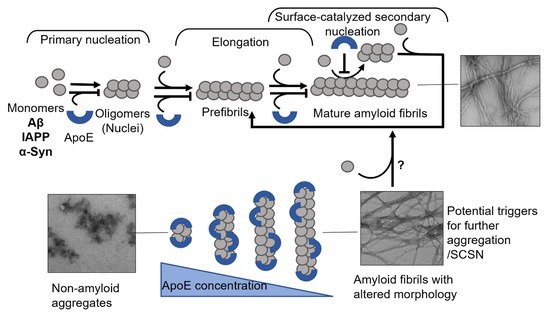
[96][135] (
Figure 4). This effect is pronounced even at highly substoichiometric ratios of ApoE:Aβ with low nM concentrations of ApoE and about 1000 times excess of Aβ. This finding indicates that ApoE most likely binds to Aβ assemblies rather than monomers (
Figure 4).
Figure 4. Mechanisms of ApoE interference with amyloid fibril formation. The schematic presentation in the upper panel shows that ApoE can target multiple steps of amyloid formation, including primary nucleation, elongation, and secondary nucleation. The lower panel shows the effect of ApoE concentration in interfering with amyloid formation process, where the higher concentration can convert the amyloid protein into non-amyloid aggregates and the lower concentrations lead to the production of amyloid fibrils with altered morphology.
Besides Aβ, only a few studies examined interactions of ApoE with other amyloid-forming proteins
[115][116][117][154,155,156]. Lei et al., showed earlier that ApoE can directly interact with IAPP in vitro, and the effect on amyloid formation is dependent on ApoE concentration, with higher concentrations promoting IAPP aggregation and lower concentrations inhibiting it
[115][154]. In contrast to this, our recent studies indicate that ApoE efficiently suppresses IAPP amyloid formation in a concentration-dependent manner starting already at highly sub-stochiometric concentrations. Upon increasing ApoE concentration in the reaction,
reswe
archers observed the formation of mainly ThT-negative assemblies with amorphous morphology
[117][156]. Interestingly,
in researchersthis study, we saw no difference in amyloid interfering ability between the three ApoE isoforms. However,
researcherswe showed that ApoE protects the cells from IAPP-induced toxicity with the weakest effect observed in the ApoE4 variant. The difference between the protective effects of ApoE isotypes is especially pronounced at low nM concentrations and longer treatment time, where ApoE2 and ApoE3 still efficiently protect the cells, but where ApoE4 almost completely loses this ability
[117][156].
Another possible target of ApoE is α-synuclein, a protein involved in dementia with Lewy bodies, including PD. However, the existing data are scarce and controversial. Gallardo et al., showed that overexpression of human α-synuclein in transgenic mice induces neurodegeneration and leads to a massive increase of ApoE and Aβ
[118][157]. While this could be a defensive response directed to reduce α-syn aggregation and protect from PD, the authors showed that the elevation of ApoE levels is accompanied by Aβ aggregation
[118][157]. Another study showed that at very low concentrations ApoE can promote α-synuclein aggregation, while higher concentrations have an inhibitory effect on the aggregation process
[116][155].
These and our data
[96][117][135,156] indicate the importance of the concentration factor of an amyloid inhibiting protein, where simply insufficient inhibition could lead to the formation of more pathological species (see
Figure 4, where in the presence of lower concentrations of ApoE, the generated fibrils adopt a more diffuse morphology) and trigger further aggregation of an amyloidogenic protein. In response to early amyloid formation, cells could possibly increase ApoE expression as a protective measure to halt the aggregation process. Failure in raising ApoE levels could then lead to a sustained amyloid formation process. Carrying the ApoE4 variant additionally increases this risk due to its intrinsic properties and weaker protective ability, which can be observed on cellular models
[73][117][112,156].
Several ApoE-based therapeutic approaches have been suggested particularly for AD and summarized in a recent review
[119][158]. Among these approaches are gene editing and gene therapy directed to recalibrate ApoE function via CRISPR-mediated or adeno-associated virus-mediated gene delivery. By those methods, it is possible to edit
ApoE4 allele to
ApoE3, replace
ApoE4 by protective
ApoE2 or overexpress
ApoE2 in
ApoE4 carriers, which could play a compensatory role for loss of function
ApoE4. Other suggested approaches include restoring or tuning the function of ApoE via enhanced lipidation or targeted modification of
ApoE4 structure. Alteration of ApoE levels is also considered as a potential disease-modifying therapy
[119][158].