 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Victoria Serelli-Lee | -- | 2450 | 2022-04-28 09:30:23 | | | |
| 2 | Rita Xu | -15 word(s) | 2435 | 2022-04-28 10:05:49 | | | | |
| 3 | Victoria Serelli-Lee | Meta information modification | 2435 | 2022-04-28 10:11:13 | | |
Video Upload Options
Biomarker-driven drug development in age of personalized medicines. A biomarker life cycle is broken down into 3 stages - discovery, translation, and qualification. Researchers review current development strategies and technologies applied at each of these stages, with emphasis on the use of real-world data as an important source of supporting evidence.
1. Introduction
The aim in drug development is to validate a clinically efficacious dose of a new drug for a defined disease state, i.e., finding the dose that will be effective for a defined group of patients while causing the least side effects. In drug development today, where a large proportion of new drug entities are targeted agents with specific molecular targets, the interpretation of this aim should be re-defined and the approach to drug development transformed. There are two parts to this transformation:
“Clinically efficacious dose”- it is well established that there are individual differences that lead to differences in clinical efficacy and severity of side effects caused by drugs. In oncology, clinical biomarkers are used to identify individuals who may respond better to targeted therapies. Pharmacogenomic (PGx) markers are also widely used now to determine dosages or identify patients who may have an adverse side effect to a drug.
“Defined disease state” - in May 2017, the Food and Drug Administration (FDA) approved pembrolizumab for microsatellite instability-high (MSI-H) or mismatch re-pair deficient (dMMR) solid tumors – the first regulatory approval of an indication defined by biomarkers[1]. This signified the beginning of biomarker-based diagnoses in the clinic.
Biomarker-driven drug development embeds knowledge of disease etiology into clinical trial designs with the aim of de-risking the process and improving success rates [2][3][4]. Over the past two decades, with advances in biotechnology, drug development and healthcare has greatly advanced especially in the field of genomic and precision medicine. Specifically, in recent years comprehensive genome profiling (CGP) using next-generation sequencing (NGS) has provided the ability to molecularly profile cancers. This provides information on the complexity of the disease and potentially identifies actionable mutations to which available targeted therapies can be prescribed.
Using an array of next-generation technologies, biomarker assays today can generate high-resolution data that provide biological information down to the single-cell level [5][6][7][8]. Accumulating omics data, clinical data, electronic health records and wearable device data are contributing to the “data overload”[9] in healthcare. The ability to apply analytics and artificial intelligence (AI) to mine this real-world data (RWD) for insights is accelerating the progress of understanding of various disease etiologies and drug interactions [10], and is changing the way researchers approach science and drug development.
2. Overview
The current state of biomarker-driven drug development can be summarized as a “Personalized therapy ecosystem” (PTE) and is represented pictorially in Figure 1. The PTE was conceived based on examples of the use of RWD/evidence (RWD/E) in the process of drug development, which will be reviewed in the following sections.
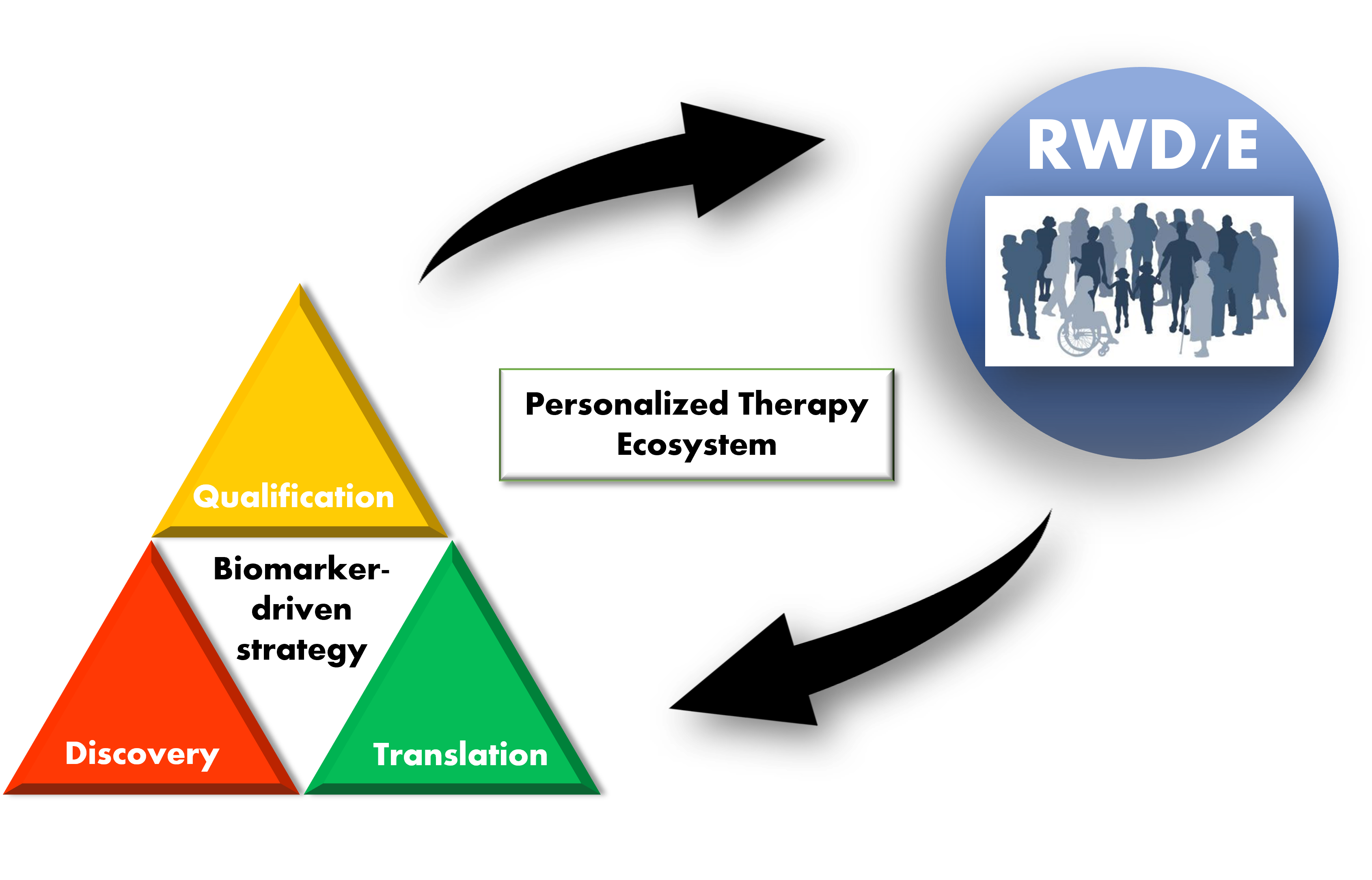
Figure 1. Personalized therapy ecosystem (PTE). A pictorial representation of a PTE. A biomarker “lifecycle” is broken down into 3 stages – discovery, translation, and qualification. Biomarker testing in the clinic generates real-world data/evidence (RWD/E) that feeds back into (supports) all stages of biomarker-driven drug development. Increased efficiency in development leads to more clinically validated biomarkers that can be used in clinical practice.
3. Definitions
“Clinical biomarkers”, as defined in the BEST (Biomarkers, EndpointS and other Tools)[11] resource, put together by the FDA-National Institutes of Health (NIH) Biomarker working group, is “a defined characteristic that is measured as an indicator of normal biological processes, pathogenic processes, or responses to an exposure or intervention, including therapeutic interventions. Molecular, histologic, radiographic, or physiologic characteristics are types of biomarkers. A biomarker is not an assessment of how an individual feels, functions, or survives”. In the PTE, a biomarker refers to all quanta of measurements - from single molecular targets to whole genome sequence, to multiplex biomarker algorithms, to omics-signatures, to high resolution medical images and digital biomarkers.
A wholistic biomarker assessment of an individual’s disease state as a disease blueprint, which is defined as the true omni-level etiology of an individual’s disease state. Like an architectural blueprint provides all the details required to construct a building, a disease blueprint reveals the pathophysiology of a disease. Depending on the disease, this blueprint could be derived via the integration of various types of biomarker information - next-generation multi-omic assay data, medical images, and even ambulatory vital sign measurements. With this blueprint at hand, targeted therapies can be accurately prescribed to patients. In the management of chronic diseases, the blueprint may also guide treatment strategies, ensuring optimization.
The concept of “personalized approach” or “right drug to right patient” takes on several nomenclatures in the literature - “targeted therapy”, “precision medicine” and “personalized medicine”. In the PTE, the term “personalized therapy” refers to a therapeutic method or drug prescribed to a patient based on their disease blueprint.
An “ecosystem” typically refers to an interconnected network. In biology, it is a network of interdependent living organisms. In the PTE, all stakeholders in drug development—drug makers, in vitro diagnostic (IVD) makers, biotechnology industry, healthcare industry and supporting infrastructure, regulatory agencies, academia, patients, and advocacy groups—are participating members. All members are interdependent for the unified benefit of eventually delivering personalized therapies to patients.
4. Biomarker Discovery
Biomarker discovery ties in closely with new drug discovery. Identifying biologically relevant and druggable targets for diseases is a key challenge in drug development. Evidence shows that deep understanding of disease pathophysiology is an important factor of success [12][13][14].
4.1. Big Data and Computational Approaches
Large-scale, nationwide initiatives to sequence the genomes of large populations provide a valuable data source to mine for research and target identification. Examples include government-led initiatives such as the 100,000 Genomes Project (UK) [15] and the million-genome Precision Medicine Initiative (US). In Japan, the Tohoku Medical Megabank (ToMMo) Project is the largest cohort, comprising clinical, genomic and multi-omics data from more than 150,000 participants [16].
Genomic analyses such as genome-wide association studies (GWAS) are traditionally conducted on large cohort data to identify quantitative trait loci in specific populations, which may shed insight into disease mechanisms. More recently, GWAS is further combined with functional genomic strategies to shed further insight into disease pathology [17]. Disease-specific molecular profiles have been identified in publicly available databases, and such insights are used for discovery, repurposing, and development [18][19].
Utilizing omics analyses and databases, biological networks can be mathematically modeled, enabling quantitative analyses of normal biological regulation versus dysfunction in pathophysiological states. Multi-scaled quantitative systems pharmacology (QSP)- models of networks involved in disease progression can be potentially used to define a wholistic profile of a disease – or a disease blueprint. Well-established models could potentially also provide the ability to predict the effects of a drug candidate on pathophysiology [20][21], thus aiding in the optimization process. Most recently, the mapping of the human interactome (genome-proteome interactions) has reached an extensive level of 53,000 high-quality protein-protein interactions [22]. The application of network science on such models can transform the way biomarkers are discovered and even the way diseases are defined [23].
Increases in the utilization and mining of such curated data may offer invaluable insights into population-specific, disease outcome-related traits, and these should be leveraged where and when possible.
4.2. Experimental Approaches
Application of patient derived cells in in vitro studies and in vivo patient-derived xenograft models is a powerful method for evaluating and optimizing drug candidates [24][25]. Human-induced pluripotent stem cells (hiPSCs), particularly when differentiated into three-dimensional multicellular models such as spheroids or organoids, may be used as models to mimic in vivo pathophysiology and pharmacological responsiveness. CRISPR/Cas9-based gene editing is more specific and powerful than traditional knockout methods in producing loss-of-function phenotypes and is now being employed to create isogenic disease models for drug screening [26] and target identification [27]. By integrating the CRISPR/Cas9 screening output with other lines of data, including real world patient data, a quantitative framework was generated for screening drug candidates. This example demonstrated the value of combining experimental and computational data-driven approaches [27].
5. Biomarker Translation
Translational research involves bringing scientific discoveries or basic research concepts into the clinic. In this section the processes involved in taking a biomarker from the R&D phase to its use in a clinical setting, including the technical assay validation process is reviewed. The BEST resource 2020 states that validation is important for ensuring that the test, tool or instrument is adequate for its proposed use [11]. Validation of a biomarker in drug development comprises of 2 main pillars:
(1) Analytical validation of the assay methodology to evaluate performance characteristics such as precision, accuracy, specificity, selectivity, sensitivity, analytical range, interference, and sample stability to broadly name a few [28][29].
(2) Clinical validation/qualification to demonstrate the relationship of a biomarker with the clinical outcome it is posited to be associated with.
The spectrum of biomarkers used in drug development spans a wide range of definitions and intended applications (proposed use) frequently also referred to as context of use (COU). The required rigor of analytical validation should correspond to the clinical or regulatory risk associated with the COU of the biomarker.
In a companion diagnostic (CDx) development program, determining the clinically relevant threshold, sometimes known as “cut-point” or “cutoff” is a key requirement in clinical validation. This threshold value should be clinically relevant in the appropriate patient population [30]. The FDA, recognizing that there is wealth of real-world clinical lab data that can be potentially utilized as evidence to support the development of IVDs, issued two guidance’s - in August 2017, “Use of RWE to Support Regulatory Decision Making for Medical Devices” [31] and then in April 2018, “Use of Public Human Genetic Variant Databases to Support Clinical Validity for Genetic and Genome-based in vitro diagnostics”[32]. A report was also recently published by the Medical Device Innovation Consortium on using RWE in pre- and post-market regulatory decision making for IVDs [33]. A well-cited example of the use of RWD in supporting clinical performance validation is the MSK-IMPACT cancer panel [34].
6. Biomarker Qualification
The “Qualification” of a biomarker demonstrates the clinical utility of the biomarker. “Clinical utility” as defined by the National Cancer Institute’s (NCI) dictionary of genetic terms is the “likelihood that a test will, by prompting an intervention, result in an improved health outcome”.
6.1. Mendelian Randomization
Mendelian randomization (MR) is an instrumental variable approach widely used in observational studies to strengthen causal inferences made retrospectively from da-ta. It is analogous to a randomized controlled trial, except that in MR, a germline genetic variant that is known to be strongly associated with an exposure, is utilized as a proxy for the risk factor of interest [35]. Mendelian randomization has been widely used in correlating disease risk factors with biomarkers [36][37] and is used to aid in the identification of potential drug targets or biomarkers.
6.2. Single Biomarker-driven Clinical Trials
Pivotal clinical trials evaluating the use of a single-biomarker CDx with a therapeutic product typically employ an “enrichment design” or a “biomarker-based strategy design”.
“Enrichment designs” enroll only biomarker-positive patients to evaluate the safety and efficacy of the treatment in the biomarker-positive population. This design is evident in the pivotal trial for trastuzumab in HER2-positive breast cancer patients [38]. Although efficient if the biomarker correlates well with the disease etiology, this approach raises the question of whether the investigational drug may also be potentially effective in the biomarker-negative population, since data from this group of subjects are not obtained in the study.
“Biomarker-based strategy design” randomizes subjects to either a biomarker-based strategy arm or a control arm (non-biomarker-based strategy). The biomarker-based strategy arm is further sub-divided into the biomarker-positive group, receiving the investigational drug, while the biomarker-negative group receives standard of care. In the control arm, subjects are assigned to the standard of care treatment. Some trial designs may also include a sub-group within the control arm receiving an investigational drug [39].
6.3. Master Protocols and Adaptive Trial Designs
Platform, basket, and umbrella trials are typical formats for a “master protocol”. Initially designed and used in oncology trials, this design enabled the simultaneous evaluation of multiple therapeutic drugs across multiple biomarker-defined populations in a single clinical trial infrastructure [40][41][42][43]. This strategy is now being employed in therapeutic areas such as chronic pain management [44], autoimmune dysfunctions [45] and COVID-19 [46], to name a few.
Bayesian adaptive trial design is used in evaluating personalized therapies in a range of diseases [47][48][49][50]. This design is an adaptive approach which allows for real time outcomes to influence ongoing treatment assignment probabilities, contributing to enhanced flexibility and efficiency. By defining early stopping rules, ineffective treatments can be terminated earlier, such that more patients can be subsequently treated with effective treatments.
6.4. Evaluating Biomarkers in Personalized Approaches
Many “precision medicine trials” in oncology have already been conducted or are ongoing. These trials molecularly profile patients’ tumor evaluate genomic, transcriptomic, or proteomic makeup and prescribes a treatment regime based on this. The I-PREDICT study [51] described that in heterogenous cancers harboring multiple genetic aberrations that have “matched therapies”, the administration of a combination of the matched therapies resulted in better patient outcomes.
In Japan, the National Cancer Center had also initiated a personalized medicine trial [52] to evaluate the clinical utility of performing a CGP panel at the time of initial diagnosis of patients with solid tumors, as compared to the current standard of performing a CGP test only after completion of current standard of care.
Even while the science may support the motive to shift toward a personalized approach, from the patient and government perspectives, assessing the value and cost effectiveness of the approach is yet another complex issue. Cost-effectiveness studies in specific disease settings may provide the needed insight into the value of personalized therapies [53][54][55][56]. Cost effectiveness and value of personalized approaches involving more complex and innovative biotechnologies are also still yet to be studied in-depth. As newer approaches are available and clinical utility validated, the ability to evaluate personalized approaches with respect to standard of care will be important in translating the method from a hypothetical to a practical one.
On top of just genomic data, multiple modes of biomarker information should ideally be comprehensively interpreted to diagnose a patient based on their disease blueprint.
7. Future Prospects
Studies that demonstrate value in biomarker-focused or personalized approaches over current standard of care, are currently lacking. It is important for stakeholders including key opinion leaders, government agencies, and regulatory bodies to be aligned on current unmet medical needs and agree on how biomarker-driven approaches can (or cannot) help address these needs. A framework for defining and evaluating “clinical benefit”, should be established, perhaps for each disease setting. This will instill more clarity and assurance on the direction that the industry should take in R&D efforts.
References
- FDA grants accelerated approval to pembrolizumab for first tissue/site agnostic indication . Food & Drug Administration. Retrieved 2022-4-28
- Richard Frank; Richard Hargreaves; Clinical biomarkers in drug discovery and development. Nature Reviews Drug Discovery 2003, 2, 566-580, 10.1038/nrd1130.
- Brett A English; Kelan Thomas; Jack Johnstone; Adam Bazih; Lev Gertsik; Larry Ereshefsky; Use of translational pharmacodynamic biomarkers in early-phase clinical studies for schizophrenia. Biomarkers in Medicine 2014, 8, 29-49, 10.2217/bmm.13.135.
- Jeffrey Cummings; The Role of Biomarkers in Alzheimer’s Disease Drug Development. Advances in Experimental Medicine and Biology 2019, 1118, 29-61, 10.1007/978-3-030-05542-4_2.
- Byungjin Hwang; Ji Hyun Lee; Duhee Bang; Single-cell RNA sequencing technologies and bioinformatics pipelines. Experimental & Molecular Medicine 2018, 50, 1-14, 10.1038/s12276-018-0071-8.
- Efrat Shema; Bradley E. Bernstein; Jason D. Buenrostro; Single-cell and single-molecule epigenomics to uncover genome regulation at unprecedented resolution. Nature Genetics 2018, 51, 19-25, 10.1038/s41588-018-0290-x.
- Jun Wang; Fan Yang; Emerging Single-Cell Technologies for Functional Proteomics in Oncology. Expert Review of Proteomics 2016, 13, 805-815, 10.1080/14789450.2016.1215920.
- Yanling Song; Xing Xu; Wei Wang; Tian Tian; Zhi Zhu; Chaoyong Yang; Single cell transcriptomics: moving towards multi-omics. The Analyst 2019, 144, 3172-3189, 10.1039/c8an01852a.
- Sabyasachi Dash; Sushil Kumar Shakyawar; Mohit Sharma; Sandeep Kaushik; Big data in healthcare: management, analysis and future prospects. Journal of Big Data 2019, 6, 54, 10.1186/s40537-019-0217-0.
- J. Gray Camp; Randall Platt; Barbara Treutlein; Mapping human cell phenotypes to genotypes with single-cell genomics. Science 2019, 365, 1401-1405, 10.1126/science.aax6648.
- BEST (Biomarkers, EndpointS, and other Tools) Resource . NIH-NLM NCBI. Retrieved 2022-4-28
- Matthew R Nelson; Hannah Tipney; Jeffery Painter; Judong Shen; Paola Nicoletti; Yufeng Shen; Aris Floratos; Pak Chung Sham; Mulin Jun Li; Junwen Wang; et al.Lon R CardonJohn WhittakerPhilippe Sanseau The support of human genetic evidence for approved drug indications. Nature Genetics 2015, 47, 856-860, 10.1038/ng.3314.
- Paul Morgan; Dean G. Brown; Simon Lennard; Mark J. Anderton; J. Carl Barrett; Ulf Eriksson; Mark Fidock; Bengt Hamrén; Anthony Johnson; Ruth E. March; et al.James MatchamJerome MettetalDavid J. NichollsStefan PlatzSteve ReesMichael A. SnowdenMenelas N. Pangalos Impact of a five-dimensional framework on R&D productivity at AstraZeneca. Nature Reviews Drug Discovery 2018, 17, 167-181, 10.1038/nrd.2017.244.
- Shuang S. Wu; Kathy Fernando; Charlotte Allerton; Kathrin U. Jansen; Michael S. Vincent; Mikael Dolsten; Reviving an R&D pipeline: a step change in the Phase II success rate. Drug Discovery Today 2020, 26, 308-314, 10.1016/j.drudis.2020.10.019.
- Vivien Marx; The DNA of a nation. Nature 2015, 524, 503-505, 10.1038/524503a.
- Soichi Ogishima; Satoshi Nagaie; Satoshi Mizuno; Ryosuke Ishiwata; Keita Iida; Kazuro Shimokawa; Takako Takai-Igarashi; Naoki Nakamura; Sachiko Nagase; Tomohiro Nakamura; et al.Naho TsuchiyaNaoki NakayaKeiko MurakamiFumihiko UenoTomomi OnumaMami IshikuroTaku ObaraShunji MugikuraHiroaki TomitaAkira UrunoTomoko KobayashiAkito TsuboiShu TadakaFumiki KatsuokaAkira NaritaMika SakuraiSatoshi MakinoGen TamiyaYuichi AokiRitsuko ShimizuIkuko N. MotoikeSeizo KoshibaNaoko MinegishiKazuki KumadaTakahiro NobukuniKichiya SuzukiInaho DanjohFuji NagamiKozo TannoHideki OhmomoKoichi AsahiAtsushi ShimizuAtsushi HozawaShinichi KuriyamaMasayuki YamamotoMichiaki AbeYayoi AizawaKoichi ChidaShinichi EgawaAi EtoTakamitsu FunayamaNobuo FuseYohei HamanakaYuki HaradaHiroaki HashizumeShinichi HiguchiSachiko HiranoTakumi HirataMasahiro HiratsukaKazuhiko IgarashiJin InoueNoriko IshidaNaoto IshiiTadashi IshiiKiyoshi ItoSadayoshi ItoMaiko KageyamaHiroshi KawameJunko KawashimaMasahiro KikuyaKengo KinoshitaKazuyuki KitataniTomomi KiyamaHideyasu KiyomotoEiichi KodamaMana KogureKaname KojimaSachie KoreedaShihoko KoyamaHisaaki KudoShigeo KureMiho KurikiYoko KurokiNorihide MaikusaHiroko MatsubaraHiroyuki MatsuiHirohito MetokiTakahiro MimoriKazuharu MisawaMasako MiyashitaHozumi MotohashiMasato NagaiMasao NagasakiKeiko NakayamaIchiko NishijimaKotaro NochiokaNoriaki OhuchiGervais OlivierNoriko OsumiHiroshi OtsuAkihito OtsukiDaisuke SaigusaSakae SaitoTomo SaitoMasaki SakaidaMika Sakurai-YagetaYuki SatoYukuto SatoAtsushi SekiguchiChen-Yang ShenTomoko F. ShibataMatsuyuki ShirotaJunichi SugawaraYoichi SuzukiMakiko TairaYuji TakanoYasuyuki TakiOsamu TanabeHiroshi TanakaYukari TanakaShunsuke TeraguchiTakahiro TerakawaTeiji TominagaIchiro TsujiMasao UekiNobuo YaegashiJunya YamagishiYumi Yamaguchi-KabataChizuru YamanakaRiu YamashitaJun YasudaJunji YokozawaKazunori WakiMakoto SasakiJunko AkaiRyujin EndoAkimune FukushimaRyohei FurukawaTsuyoshi HachiyaKouhei HashizumeJiro HitomiYasushi IshigakiShohei KomakiYuka KotozakiTakahiro MikamiMotoyuki NakamuraNaoyuki NishiyaSatoshi NishizukaYoko NomuraKuniaki OgasawaraShinichi OmamaRyo OtomoKotaro OtsukaKiyomi SakataRyohei SasakiMamoru SatohNamie SatoYu ShiwaYoichi SutohNobuyuki TakanashiNoriko TakebeFumitaka TanakaRyoichi TanakaTomoharu TokutomiKayono YamamotoFumio Yamashitathe Tohoku Medical Megabank Project Study Group dbTMM: an integrated database of large-scale cohort, genome and clinical data for the Tohoku Medical Megabank Project. Human Genome Variation 2021, 8, 1-8, 10.1038/s41439-021-00175-5.
- Eddie Cano-Gamez; Gosia Trynka; From GWAS to Function: Using Functional Genomics to Identify the Mechanisms Underlying Complex Diseases. Frontiers in Genetics 2020, 11, 424, 10.3389/fgene.2020.00424.
- Yadi Zhou; Jiansong Fang; Lynn M. Bekris; Young Heon Kim; Andrew A. Pieper; James B. Leverenz; Jeffrey Cummings; Feixiong Cheng; AlzGPS: a genome-wide positioning systems platform to catalyze multi-omics for Alzheimer’s drug discovery. Alzheimer's Research & Therapy 2021, 13, 1-13, 10.1186/s13195-020-00760-w.
- E. L. Leung; Z.-W. Cao; Z.-H. Jiang; H. Zhou; L. Liu; Network-based drug discovery by integrating systems biology and computational technologies. Briefings in Bioinformatics 2012, 14, 491-505, 10.1093/bib/bbs043.
- Violeta Balbas-Martinez; Leire Ruiz-Cerdá; Itziar Irurzun-Arana; Ignacio González-García; An Vermeulen; José David Gómez-Mantilla; Iñaki F. Trocóniz; A systems pharmacology model for inflammatory bowel disease. PLoS ONE 2018, 13, e0192949, 10.1371/journal.pone.0192949.
- Kirill Peskov; Ivan Azarov; Lulu Chu; Veronika Voronova; Yuri Kosinsky; Gabriel Helmlinger; Quantitative Mechanistic Modeling in Support of Pharmacological Therapeutics Development in Immuno-Oncology. Frontiers in Immunology 2019, 10, 924, 10.3389/fimmu.2019.00924.
- Katja Luck; Dae-Kyum Kim; Luke Lambourne; Kerstin Spirohn; Bridget E. Begg; Wenting Bian; Ruth Brignall; Tiziana Cafarelli; Francisco José Campos Laborie; Benoit Charloteaux; et al.Dongsic ChoiAtina G. CotéMeaghan DaleySteven DeimlingAlice DesbuleuxAmélie DricotMarinella GebbiaMadeleine F. HardyNishka KishoreJennifer J. KnappIstvan KovacsIrma LemmensMiles W. MeeJoseph C. MellorCarl PollisCarles PonsAaron D. RichardsonSadie SchlabachBridget TeekingAnupama YadavMariana BaborDawit BalchaOmer BashaChristian Bowman-ColinSuet-Feung ChinSoon Gang ChoiClaudia ColabellaGeorges CoppinCassandra D’AmataDavid De RidderSteffi De RouckMiquel Duran-FrigolaHanane EnnajdaouiFlorian GoebelsLiana GoehringAnjali GopalGhazal HaddadElodie HatchiMohamed HelmyYves JacobYoseph KassaSerena LandiniRoujia LiNatascha van LieshoutAndrew MacWilliamsDylan MarkeyJoseph N. PaulsonSudharshan RangarajanJohn RaslaAshyad RayhanThomas RollandAdriana San-MiguelYun ShenDayag SheykhkarimliGloria M. SheynkmanEyal SimonovskyMurat TaşanAlexander TejedaVincent TropepeJean-Claude TwizereYang WangRobert WeatherittJochen WeileYu XiaXinping YangEsti Yeger-LotemQuan ZhongPatrick AloyGary D. BaderJavier De Las RivasSuzanne GaudetTong HaoJanusz RakJan TavernierDavid E. HillMarc VidalFrederick P. RothMichael A. Calderwood A reference map of the human binary protein interactome. Nature 2020, 580, 402-408, 10.1038/s41586-020-2188-x.
- Albert-László Barabási; Zoltán N. Oltvai; Network biology: understanding the cell's functional organization. Nature Reviews Microbiology 2004, 5, 101-113, 10.1038/nrg1272.
- Ian R. Powley; Meeta Patel; Gareth Miles; Howard Pringle; Lynne Howells; Anne Thomas; Catherine Kettleborough; Justin Bryans; Tim Hammonds; Marion Macfarlane; et al.Catrin Pritchard Patient-derived explants (PDEs) as a powerful preclinical platform for anti-cancer drug and biomarker discovery. British Journal of Cancer 2020, 122, 735-744, 10.1038/s41416-019-0672-6.
- Jong-Chan Park; So-Yeong Jang; Dongjoon Lee; Jeongha Lee; Uiryong Kang; Hongjun Chang; Haeng Jun Kim; Sun-Ho Han; Jinsoo Seo; Murim Choi; et al.Dong Young LeeMin Soo ByunDahyun YiKwang-Hyun ChoInhee Mook-Jung A logical network-based drug-screening platform for Alzheimer’s disease representing pathological features of human brain organoids. Nature Communications 2021, 12, 1-13, 10.1038/s41467-020-20440-5.
- Courtney S. Young; Michael R. Hicks; Natalia V. Ermolova; Haruko Nakano; Majib Jan; Shahab Younesi; Saravanan Karumbayaram; Chino Kumagai-Cresse; Derek Wang; Jerome A. Zack; et al.Donald B. KohnAtsushi NakanoStanley F. NelsonM. Carrie MiceliMelissa J. SpencerApril D. Pyle A Single CRISPR-Cas9 Deletion Strategy that Targets the Majority of DMD Patients Restores Dystrophin Function in hiPSC-Derived Muscle Cells. Cell Stem Cell 2016, 18, 533-540, 10.1016/j.stem.2016.01.021.
- Fiona M. Behan; Francesco Iorio; Gabriele Picco; Emanuel Gonçalves; Charlotte M. Beaver; Giorgia Migliardi; Rita Santos; Yanhua Rao; Francesco Sassi; Marika Pinnelli; et al.Rizwan AnsariSarah HarperDavid Adam JacksonRebecca McRaeRachel PooleyPiers WilkinsonDieudonne van der MeerDavid DowCarolyn Buser-DoepnerAndrea BertottiLivio TrusolinoEuan A. StronachJulio Saez-RodriguezKosuke YusaMathew J. Garnett Prioritization of cancer therapeutic targets using CRISPR–Cas9 screens. Nature 2019, 568, 511-516, 10.1038/s41586-019-1103-9.
- Steven Piccoli; Devangi Mehta; Alessandra Vitaliti; John Allinson; Shashi Amur; Steve Eck; Cherie Green; Michael Hedrick; Shirley Hopper; Allena Ji; et al.Alison JoyceVirginia LitwinKevin MaherJoel MathewsKun PengAfshin SafaviYow-Ming WangYan ZhangLakshmi AmaravadiNisha PalackalSai ThankamonyChris BeaverEris BameThomas EmrichChristine GrimaldiJonathan HaulenbeekVellalore KakkanaiahDavid LanhamAndrew MayerPaul C TrampontLaurent VermetNaveen DakappagariCatherine FleenerFabio GarofoloCynthia RogersShabnam TangriYuanxin XuMeina LiangManoj RajadhyakshaSusan RichardsBecky SchweighardtShobha PurushothamaDaniel BaltrukonisJochen BrummElana CherryJason DelcarpiniCarol GleasonSusan KirshnerRobert KubiakLuying PanMichael PartridgeJoão Pedras-VasconcelosQiang QuVenke SkibeliTherese Solstad SaundersRoland F StaackKay StubenrauchAl TorriDaniela VerthelyiHaoheng YanBoris GorovitsRachel PalmerMark MiltonBrian LongBart CorsaroVahid FarrokhiMichele FiscellaNeil HendersonVibha JawaJim McNallyRocio MurphyHanspeter WaldnerTong-Yuan Yang 2019 White Paper on Recent Issues in Bioanalysis: FDA Immunogenicity Guidance, Gene Therapy, Critical Reagents, Biomarkers and Flow Cytometry Validation (Part 3 – Recommendations on 2019 FDA Immunogenicity Guidance, Gene Therapy Bioanalytical Challenges, Strategies for Critical Reagent Management, Biomarker Assay Validation, Flow Cytometry Validation & CLSI H62). Bioanalysis 2019, 11, 2207-2244, 10.4155/bio-2019-0271.
- Jean W. Lee; Viswanath Devanarayan; Yu Chen Barrett; Russell Weiner; John Allinson; Scott Fountain; Stephen Keller; Ira Weinryb; Marie Green; Larry Duan; et al.James A. RogersRobert MillhamPeter J. O'brienJeff SailstadMasood KhanChad RayJohn A. Wagner Fit-for-Purpose Method Development and Validation for Successful Biomarker Measurement. Pharmaceutical Research 2006, 23, 312-328, 10.1007/s11095-005-9045-3.
- Edward B. Garon; Naiyer A. Rizvi; Rina Hui; Natasha Leighl; Ani S. Balmanoukian; Joseph Paul Eder; Amita Patnaik; Charu Aggarwal; Matthew Gubens; Leora Horn; et al.Enric CarcerenyMyung-Ju AhnEnriqueta FelipJong-Seok LeeMatthew D. HellmannOmid HamidJonathan W. GoldmanJean-Charles SoriaMarisa Dolled-FilhartRuth Z. RutledgeJin ZhangJared K. LuncefordReshma RangwalaGregory M. LubinieckiCharlotte RoachKenneth EmancipatorLeena Gandhi Pembrolizumab for the Treatment of Non–Small-Cell Lung Cancer. New England Journal of Medicine 2015, 372, 2018-2028, 10.1056/nejmoa1501824.
- Use of Real-World Evidence to Support Regulatory Decision-Making for Medical Devices . Center for Devices and Radiological Health. Retrieved 2022-4-28
- Use of Public Human Genetic Variant Databases to Support Clinical Validity for Genetic and Genomic-Based In Vitro Diagnostics . Center for Devices and Radiological Health. Retrieved 2022-4-28
- Real-World Clinical Evidence Generation: Advancing Regulatory Science and Patient Access for In Vitro Diagnostics (IVDs) . Medical Device Innovation Consortium. Retrieved 2022-4-28
- FDA: MSK-IMPACT Decision Summary . Food & Drug Administration. Retrieved 2022-4-28
- Debbie A. Lawlor; Roger M. Harbord; Jonathan A. C. Sterne; Nic Timpson; George Davey Smith; Mendelian randomization: Using genes as instruments for making causal inferences in epidemiology. Statistics in Medicine 2008, 27, 1133-1163, 10.1002/sim.3034.
- Zhao Yang; C. Mary Schooling; Man Ki Kwok; Mendelian randomization study of interleukin (IL)-1 family and lung cancer. Scientific Reports 2021, 11, 1-11, 10.1038/s41598-021-97099-5.
- Susanna C Larsson; Matthew Traylor; Rainer Malik; Martin Dichgans; Stephen Burgess; Hugh S Markus; Modifiable pathways in Alzheimer’s disease: Mendelian randomisation analysis. BMJ 2017, 359, j5375, 10.1136/bmj.j5375.
- W. Eiermann; Trastuzumab combined with chemotherapy for the treatment of HER2-positive metastatic breast cancer: Pivotal trial data. Annals of Oncology 2001, 12, S57-S62, 10.1093/annonc/12.suppl_1.s57.
- Boris Freidlin; Edward L. Korn; Biomarker enrichment strategies: matching trial design to biomarker credentials. Nature Reviews Clinical Oncology 2013, 11, 81-90, 10.1038/nrclinonc.2013.218.
- Vladimir Bogin; Master protocols: New directions in drug discovery. Contemporary Clinical Trials Communications 2020, 18, 100568, 10.1016/j.conctc.2020.100568.
- Jay J. H Park; Ellie Siden; Michael J. Zoratti; Louis Dron; Ofir Harari; Joel Singer; Richard T. Lester; Kristian Thorlund; Edward J. Mills; Systematic review of basket trials, umbrella trials, and platform trials: a landscape analysis of master protocols. Trials 2019, 20, 1-10, 10.1186/s13063-019-3664-1.
- Mary W. Redman; Carmen J. Allegra; The Master Protocol Concept. Seminars in Oncology 2015, 42, 724-730, 10.1053/j.seminoncol.2015.07.009.
- Janet Woodcock; Lisa M. LaVange; Master Protocols to Study Multiple Therapies, Multiple Diseases, or Both. New England Journal of Medicine 2017, 377, 62-70, 10.1056/nejmra1510062.
- A Study of LY3016859 in Participants With Osteoarthritis . ClinicalTrials.gov. Retrieved 2022-4-28
- Ulla Derhaschnig; Jim Gilbert; Ulrich Jäger; Georg Böhmig; Georg Stingl; Bernd Jilma; Combined integrated protocol/basket trial design for a first-in-human trial.. Orphanet Journal of Rare Diseases 2016, 11, 134, 10.1186/s13023-016-0494-z.
- Tom Wilkinson; Rupert Dixon; Clive Page; Miles Carroll; Gareth Griffiths; Ling-Pei Ho; Anthony De Soyza; Timothy Felton; Keir E. Lewis; Karen Phekoo; et al.James D. ChalmersAnthony GordonLorcan McGarveyJillian DohertyRobert C. ReadManu Shankar-HariNuria Martinez-AlierMichael O’KellyGraeme DuncanRoelize WallesJames SykesCharlotte SummersDave SinghOn Behalf Of The Accord Collaborators ACCORD: A Multicentre, Seamless, Phase 2 Adaptive Randomisation Platform Study to Assess the Efficacy and Safety of Multiple Candidate Agents for the Treatment of COVID-19 in Hospitalised Patients: A structured summary of a study protocol for a randomised controlled trial. Trials 2020, 21, 1-3, 10.1186/s13063-020-04584-9.
- Xian Zhou; Suyu Liu; Edward S Kim; Roy S Herbst; J Jack Lee; Bayesian adaptive design for targeted therapy development in lung cancer — a step toward personalized medicine. Clinical Trials 2008, 5, 181-193, 10.1177/1740774508091815.
- Wentian Guo; Yuan Ji; Daniel V. T. Catenacci; A subgroup cluster-based Bayesian adaptive design for precision medicine. Biometrics 2016, 73, 367-377, 10.1111/biom.12613.
- Sean P. Collins; Christopher J. Lindsell; Peter S. Pang; Alan B. Storrow; W. Frank Peacock; Phil Levy; M. Hossein Rahbar; Deborah Del Junco; Mihai Gheorghiade; Donald A. Berry; et al. Bayesian adaptive trial design in acute heart failure syndromes: Moving beyond the mega trial. American Heart Journal 2012, 164, 138-145, 10.1016/j.ahj.2011.11.023.
- Xuemin Gu; Nan Chen; Caimiao Wei; Suyu Liu; Vassiliki A. Papadimitrakopoulou; Roy S. Herbst; Jiunkae Jack Lee; Bayesian Two-Stage Biomarker-Based Adaptive Design for Targeted Therapy Development. Statistics in Biosciences 2014, 8, 99-128, 10.1007/s12561-014-9124-2.
- Jason K. Sicklick; Shumei Kato; Ryosuke Okamura; Hitendra Patel; Mina Nikanjam; Paul T. Fanta; Michael E. Hahn; Pradip De; Casey Williams; Jessica Guido; et al.Benjamin M. SolomonRana R. McKayAmy KrieSarah G. BolesJeffrey S. RossJ. Jack LeeBrian Leyland-JonesScott M. LippmanRazelle Kurzrock Molecular profiling of advanced malignancies guides first-line N-of-1 treatments in the I-PREDICT treatment-naïve study. Genome Medicine 2021, 13, 1-14, 10.1186/s13073-021-00969-w.
- Advanced Medical Care Approval for Cancer Gene Panel Testing at the Time of Initial Treatment - Prospective Study to Assess the Feasibility and Clinical Utility of Comprehensive Genomic Profiling at the Time of Initial Treatment of Patients with Solid Tumors . National Cancer Center Japan. Retrieved 2022-4-28
- Sandjar Djalalov; Jaclyn Beca; Jeffrey S. Hoch; Murray Krahn; Ming-Sound Tsao; Jean-Claude Cutz; Natasha B. Leighl; Cost Effectiveness ofEML4-ALKFusion Testing and First-Line Crizotinib Treatment for Patients With AdvancedALK-Positive Non–Small-Cell Lung Cancer. Journal of Clinical Oncology 2014, 32, 1012-1019, 10.1200/jco.2013.53.1186.
- J. J. Carlson; W. Canestaro; A. Ravelo; W. Wong; The cost-effectiveness of alectinib in anaplastic lymphoma kinase-positive (ALK+) advanced NSCLC previously treated with crizotinib. Journal of Medical Economics 2017, 20, 671-677, 10.1080/13696998.2017.1302453.
- Dong Ding; Huabin Hu; Yin Shi; Longjiang She; Linli Yao; Youwen Zhu; Shan Zeng; Liangfang Shen; Jin Huang; Cost-Effectiveness of Pembrolizumab plus Axitinib Versus Sunitinib as First-Line Therapy in Advanced Renal Cell Carcinoma in the U.S.. The Oncologist 2020, 26, e290-e297, 10.1002/onco.13522.
- Eric Jutkowitz; Maureen Dubreuil; Na Lu; Karen M. Kuntz; Hyon K. Choi; The cost-effectiveness of HLA-B*5801 screening to guide initial urate-lowering therapy for gout in the United States. Seminars in Arthritis and Rheumatism 2016, 46, 594-600, 10.1016/j.semarthrit.2016.10.009.

