Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ricardo Maccioni | -- | 2464 | 2022-04-27 18:55:53 | | | |
| 2 | Catherine Yang | Meta information modification | 2464 | 2022-04-28 03:27:22 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Maccioni, R.; , .; Singh, S.; Churruca, M. Alzheimer’s Disease and Tau Self-Assembly. Encyclopedia. Available online: https://encyclopedia.pub/entry/22398 (accessed on 24 July 2026).
Maccioni R, , Singh S, Churruca M. Alzheimer’s Disease and Tau Self-Assembly. Encyclopedia. Available at: https://encyclopedia.pub/entry/22398. Accessed July 24, 2026.
Maccioni, Ricardo, , Sandeep Singh, Macarena Churruca. "Alzheimer’s Disease and Tau Self-Assembly" Encyclopedia, https://encyclopedia.pub/entry/22398 (accessed July 24, 2026).
Maccioni, R., , ., Singh, S., & Churruca, M. (2022, April 27). Alzheimer’s Disease and Tau Self-Assembly. In Encyclopedia. https://encyclopedia.pub/entry/22398
Maccioni, Ricardo, et al. "Alzheimer’s Disease and Tau Self-Assembly." Encyclopedia. Web. 27 April, 2022.
Copy Citation
Alzheimer’s disease (AD) is a multifactorial neurodegenerative disease characterized by progressive cognitive impairment, apathy, and neuropsychiatric disorders. Two main pathological hallmarks have been described: neurofibrillary tangles, consisting of tau oligomers (hyperphosphorylated tau) and Aβ plaques. The influence of protein kinases and phosphatases on the hyperphosphorylation of tau is already known. Hyperphosphorylated tau undergoes conformational changes that promote its self-assembly.
Alzheimer’s disease
tau protein
polyanions
1. Introduction: Alzheimer’s Pathogenesis
Alzheimer’s disease (AD) is a multifactorial, neurodegenerative disease, which is characterized by progressive cognitive decline and behavioral disorders of patients. Within its prognosis, three clinical stages are recognized: (1) The hippocampal stage, where the first signs of memory and judgment or executive capacity defects are evident. (2) The parieto-temporo-occipital stage (also prefrontal), associated with moderate and more severe dementia, with language disorders and others. (3) Stage of global damage, the patient presents loss of language, dysphagia, rigidity, and prostration. Aging is one of the main risk factors for AD since most of the cases are related to elderly individuals, older than 65 years, affected by so-called “sporadic AD”, currently the most common type of dementia in the senile population [1][2].
Nowadays, 50 million people are affected with AD worldwide, and this number is estimated to grow to 82 million by 2030 and 152 million by 2050 (World Alzheimer’s Report, 2018). In the global context, the costs of dementia were USD 1 trillion in 2018; 25% of this is direct medical costs and 75% indirect social costs for caregivers. In this context, this epidemic disorder is concerning for public health, with efforts focused on its prevention and treatment.
It should be noted that one of the major challenges to developing an innovative therapy or an effective diagnostic tool for AD is precisely the fact that it is a multifactorial disease. A risk factor associated with the origin and evolution of AD is the alteration of glucose metabolism that causes insulin resistance and type II diabetes mellitus (DM-II). This is demonstrated by several epidemiological studies [3][4][5]. For example, deterioration in glucose metabolism can decrease the energy available for cognitive information processes and lead to synaptic dysfunction. This happens in patients with mild cognitive impairment. If insulin resistance is maintained for longer, mitochondrial damage may be greater and oxidative damage may increase, leading to neurodegeneration in the brain. However, the mechanisms of how insulin resistance and DM work as risk factors for AD are still uncertain [6]. Another factor related to AD is depression; there is comorbidity in 50% of cases of older people with AD. Depression is considered part of the initial symptoms or a preclinical phase [7]. A relationship between high levels of beta-amyloid with increased depressive and anxiety symptoms has been evidenced. Supporting this hypothesis, neuropsychiatric symptoms could be predictors of AD [8][9][10]. This association is relevant because several diseases resulting from physiological alteration of depression (deregulation of the hypothalamic–pituitary axis, neuroinflammation, and cerebrovascular changes) have been associated with amyloid deposition [11]. Stress events may lead to depression [12] altering the HPA axis by increasing glucocorticoid production [13]. The “damage, signal” caused by stress results in the activation of the defense system mediated by glial cells of the innate immune system [14][15]. This has been related to previous stages of AD [16] and concurs with the theory of neuroimmunomodulation [11][17].
AD is characterized by two major hallmarks: (i) senile plaques (SP), composed of deposits of the amyloid-β (Aβ) peptide of 39 to 42 aminoacidic residues, generated by the proteolytic excision of the amyloid precursor protein (APP) by β and γ secretases, in the extracellular space [18]; and (ii) neurofibrillary tangles (NFT), derived from the progressive aggregation of hyperphosphorylated tau protein, inside neurons, that derive from tau assembly into oligomeric structures named “paired helical filaments” (PHF). PHFs block transport systems in neurons, thus affecting synaptic transmission [19]. The abnormal hyperphosphorylation of tau has deleterious pathological effects which are directly involved in neurodegeneration. In this context, tau protein, with its extensive pathological role in neurodegenerative diseases, is a very a promising target for therapeutic interventions. Additionally, the previously mentioned amyloid beta-protein has been the main therapeutic target in a vast array of research and treatments in the past 20 years. Nevertheless, the results of clinical and preclinical trials have been inconclusive, showing the need to find a new treatment strategy for AD [20]. Donepezil is one of the few drugs approved for AD treatment, as it is a specific inhibitor of the AchE, and recently its high affinity for human transferrin [21] has been observed, indicating an involvement in iron homeostasis.
After clarifying the major role of tau hyperphosphorylations in AD pathogenesis, a major question was to elucidate the molecular signals “upstream” that lead to the pathological phospho-tau variant. AD is a summatory of neuroinflammatory events that result from the innate immunity phenomenon of activation of microglial cells by the so-called “damage signals” that include a large set of molecular or physical factors [22][23] (Figure 1). The neuroinflammatory response involves an over-release of proinflammatory cytokines that finally signal neuronal receptors, resulting in the formation of a stable CDK5/p25 complex responsible for part of tau hyperphosphorylations. This is the basis of the Neuroimmunomodulation theory of Alzheimers disease [1][14][17][22][23]. In essence, AD is a consequence of disturbances in the cross-talks between microglial and neuronal cells that finally renders a modified phospho-tau variant followed by misfolding of tau’s structure. Thus, the neuroimmunomodulation theory has been able to explain the mechanism underlying the molecular basis of AD, allowing us to appropriately select the targets of future therapies, as well as early biomarkers for this disease. In the neuroimmunomodulation theory, the “damage signals” (such as Aβ peptide, reactive oxygen species, iron overload, vitamin B12 deficiency, free tau fragments, etc.) activate the microglia, which switch from a quiescent state to an activated (M1) state. The M1 microglia generates proinflammatory cytokines, such as IL-6 and TNF-α, generating a proinflammatory microenvironment in the brain. These cytokines also promote the assembly of the CDK5/p25 complex, promoting the hyperphosphorylation of tau. The latter eventually leads to neurodegeneration, in which some PHF fragments can act as novel danger signals in surrounding microglia, in a cyclic event.
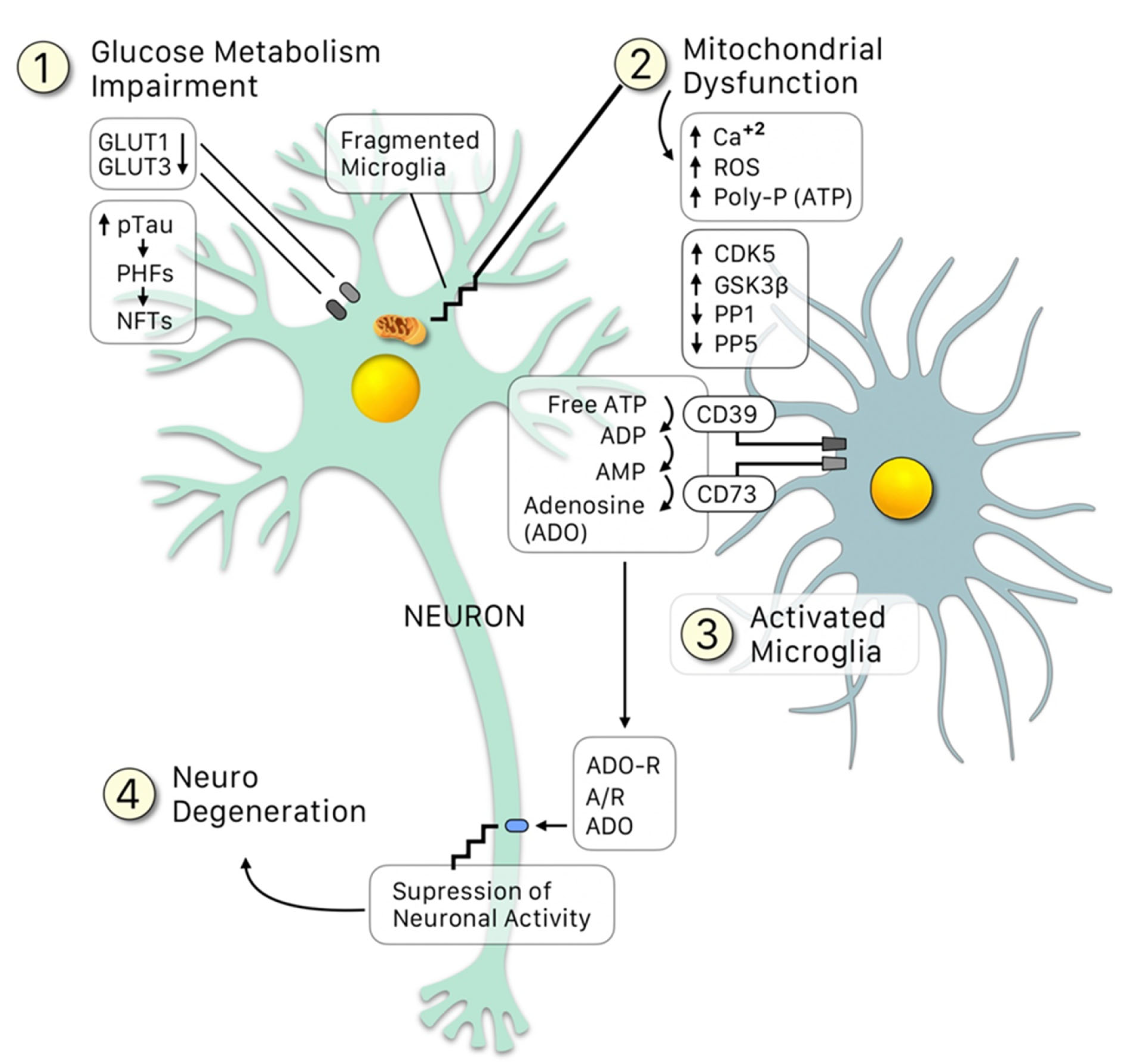
Figure 1. Crosstalk between neurons and microglia: An update of the Neuroimmunomodulation theory [2][15], in which besides the conventional “damage signals”, the glucose metabolism impairment leads to a downregulation of the GLUT1 and GLUT3 receptors, resulting in: (1) Loss of providing enough glucose for the mitochondria to function properly (2) All the latter leading to upregulation of the kinases (CDK5, GSK3β) and downregulation of the phosphatases (PP1 and PP5), which increase p-tau, generating PHF and NFT. Fragments of these damaged mitochondria, alongside NFT fragments (among other signals), activate the microglia (3). The microglia is the able to sense an increase in the free ATP (due to the fragmentation of the mitochondria) and transforms it into adenosine mediated by the clusters CD39 and CD73. Adenosine then binds to its receptor, ADO-R, over-suppressing the neuronal activity, thus leading to neurodegeneration.
2. Hyperphosphorylation of Tau: The Influence of Protein Kinases and Phosphatases during the Course of AD
Tau is a microtubule-associated protein (MAP) that is probably involved in the control of axonal transport [24]. The gene encoding tau is located in chromosome 17, region 17q 21, in humans [25]. It is structurally considered an elastic molecule and no sequential structure has been detected [26]. Its plasticity allows some of its exons (2,3,4A, 6, 8,10 and 14) to be spliced in an alternative way. This generates 6 isoforms, and it contains two major domains, an amino terminal domain and a carboxy-terminal domain. The first one is denominated ‘projection domain’, which is rich in proline and also has an acid region. The C-terminal is the principal binding domain of the microtubules and is organized by three (3R) or four (4R) internal repetitions [27].
The two main functions of tau are, under normal conditions, to provide stability to the microtubules (MT) and to articulate the transport system of signaling molecules and cellular components [20][28]. Those functions are disrupted once tau is hyperphosphorylated during the course of AD [29]. This process increases the number of binding sites in the same phosphorylated tau molecule [30]. Hyperphosphorylated tau captures native tau and other microtubule-associated proteins, causing the disassembly of microtubules [19][31].
Subsequently, there is a destabilization of the cytoskeleton, produced by the alteration of tau-dependent cellular functions, such as vesicular and organelle transport, axonal growth and nerve signal propagation [32]. This anomaly is known as tauopathy and is present in many neurodegenerative diseases [28]. There are 20 diseases categorized as tauopathies, which are sub-divided into two groups; Alzheimer Disease is part of the secondary group and it is also the most preponderant. The secondary group is characterized for the presence of both an intracellular tau pathology and an extracellular amyloid plaque deposit. The particularity of these tauopathies is the formation of insoluble deposits called neurofibrillary tangles (NFT) in the three (3R) and four (4R) isoforms. The dysfunctionality caused by NFT is manifested from the soma to the dendrites, and the most commonly affected regions of the brain are the entorhinal cortex, the hippocampus and neocortex [33].
Studies have demonstrated how an increased activity of kinases, such as CDK5, and downregulation of phosphatases, influences tau hyperphosphorylation, leading to the oligomer formation of tau [34][35][36]. The deregulation of CDK5 is due to the formation of the CDK5/p25 complex, a product of p35 splitting, possibly as a result of oxidative stress and amyloid peptides to which the neuron has been exposed. The conversion from p35 to p25 occurs through proteolysis. The result is p25, a fragment of the protein that is neurotoxic and has an active and a totally extended conformation. The conformational change suffered by p35 produces p25. Additionally, that conformational change impacts the way CDK5 activates, given the fact that the latter activation lasts longer than p35. This conversion results in CDK5 hyperactivity, and subsequently, a possible hyperphosphorylation of tau protein and neurofilaments, along with a cytoskeletal alteration and eventually neuronal death [37].
Their conformational structure changes from an α-helix to a β-sheet structure, which facilitates the formation of the oligomers. Thus, phosphorylation of tau is the main post-translational modification that is involved with AD [30].
In AD, the nature of the tau amino acids that are the target of the protein kinases is just as important as the proper hyperphosphorylation. Thus, the key amino acid phosphorylations are those critical for tau conformational changes, from an alpha-helix to a beta-sheet structure. In that regard, it was demonstrated in vitro that oxidative stress promotes tau dephosphorylation at the Tau1 epitope in SHSY5Y cells [38]. The latter was dependent on the activity of the cdk5/p35 complex, since an increase in the substrate phosphorylation as well as the complex association were observed. Additionally, oxidative stress induced a decrease in the amount of inhibitor-2 bound to phosphatase PP1, associated with an increased phosphorylation of the inhibitor-2 protein. Thus, hyperphosphorylation of tau relies on a shift of the balance between the kinases and phosphatases, in which the upregulation of the kinases’ activity exceeds the phosphatase activity.
However, there are other post-translational modifications of tau protein, besides hyperphosphorylation, that play different roles during the pathological processes leading to AD. These modifications include truncation and glycosylation; both of which occur in early stages of AD. In a study by Takahashi et al. (1999), it was observed that tau proteins were abnormally glycosylated in the brain of AD patients, which was not detected in control patients [39]. Other types of post-translational modifications are glycation, nitration, ubiquitination and polyamination [28].
Liu et al. (2002) have shown that abnormal in vitro glycosylations modulate the phosphorylation of tau by the kinases PKA, GSK-3 and CDK-5 [40], which in turn, inhibit dephosphorylation by the phosphatases PP2A and PP5 [41]. Interaction between many post-translational modifications may be necessary to induce the oligomer tau formation [30].
Additionally, the role of the microtubule regulating protein kinase MARK4 in AD has been demonstrated, as its overexpression increases tau hyperphosphorylation. In that regard, this kinase is inhibited by AChE inhibitors Donepezil and Rivastigmine [42]. Similarly, irisin, a hormone usually increased during physical exercise, has also an inhibitory potential over MARK4 [43]
Another aspect to be considered is diagnosis, as currently in AD there are several radiotracers used for positron-emission tomography (PET) that bind tau filaments, such as THK5317, THK5351, AV-1451, and PBB3 [44]. APN-1607 is one of the newest radiotracers successfully bound to PHF [45]. This is important since the design of the tracers depends on the tau structure. Specific phosphorylated tau tracers for PET scans would make the diagnosis more accurate, but their design is still a challenge.
3. Tau Auto-Aggregation and the Hypothesis of Expansion
In AD, a minimum of 30 serine/threonine residues of tau are phosphorylated, and the level of phosphorylation is correlated with the severity of the pathology. It has been demonstrated that purified phospho-tau is capable of self-polymerization in vitro without inducer molecules. Additionally, hyperphosphorylated tau can sequester tau protein from microtubules. This highlights the critical role of phosphorylation’s in tau oligomerization, and provides evidence that points to this event as one of the first milestones in AD [46]. In addition, if NFTs appear in the entorhinal cortex in the early stages of AD, and are later found in adjacent areas that are anatomically connected [47], it would mean that NFTs could be transmitted from one neuron to another [48]. So, the expansion from intracellular to extracellular is the next step. Can the PHF from the extracellular region access a nearby neuronal cell? The infectious hypothesis states that extracellular aggregates can access another cell, where they act as nucleation seeds to form larger aggregates of tau.
The formation of larger tau aggregates, together with other apoptotic signals (e.g., upregulation of suppression of neuronal activity by microglia), lead to neurodegeneration (Figure 1). Tau aggregates, in particular PHF, can be released to the extracellular once the cell enters apoptosis, where they can access a nearby cell and act as nucleation seeds for the formation of larger tau aggregates, in a cyclic process (Figure 2).
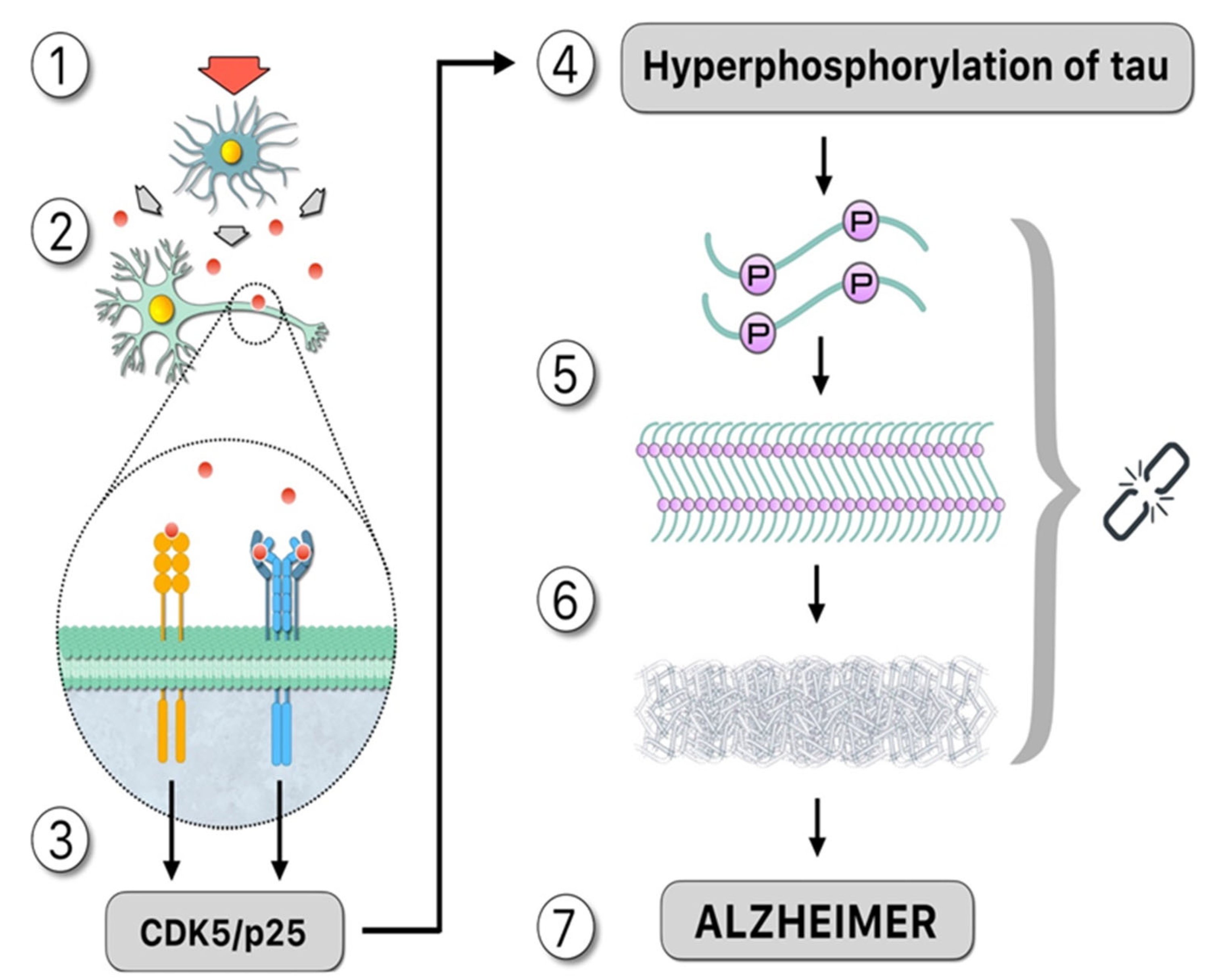
Figure 2. Upstream: “Damage signals”, represented by the red arrow, activate the microglia that release cytotoxic compounds such as cytokines, as demonstrated by the theory of neuroimmunomodulation [15]: (1). These proinflammatory cytokines, such as IL-1 and IL-6, signaled by red circles, activate membrane receptors in the neuron (2) which activates a signaling pathway that promotes the formation of the CDK5/p25 complex, which is mainly responsible for tau hyperphosphorylation (3). Downstream: Hyperphosphorylated tau increasing the number of binding sites for phosphates in the same molecule (4). Structural changes occur as a result of post-translational modifications, facilitating the formation of oligomers in neurons. Therefore, the ‘native’ protein is altered, and subsequently tau folds abnormally (misfolding) (5). Hyperphosphorylated tau, not bound to microtubules, binds in pairs, forming paired helical filaments (PHF). The polymerization of tau requires inductor molecules such as polyanions (6). Consequences of this pathological process include neural dysfunction or death from apoptosis. In this case, the PHF moves to the extracellular fluid accessing the nearby cell, acting as a nucleation seed and forming new aggregates of tau, and AD disease is observed (7). Symbology: 1.—Red arrow: damage signal. 2.—Red circle: cytokines. 3.—Gray key: hyperphosphorylation process. 4.—Chain: the missing link.
References
- Morales, I.; Cerda-Troncoso, C.; Andrade, V.; Maccioni, R.B. The Natural Product Curcumin as a Potential Coadjuvant in Alzheimer’s Treatment. J. Alzheimers Dis. 2017, 60, 451–460.
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a Common Feature of Neurodegenerative Disorders. Front. Pharm. 2019, 10, 1008.
- Hao, K.; Di Narzo, A.F.; Ho, L.; Luo, W.; Li, S.; Chen, R.; Li, T.; Dubner, L.; Pasinetti, G.M. Shared genetic etiology underlying Alzheimer’s disease and type 2 diabetes. Mol. Asp. Med. 2015, 43, 66–76.
- Matsuzaki, T.; Sasaki, K.; Tanizaki, Y.; Hata, J.; Fujimi, K.; Matsui, Y.; Sekita, A.; Suzuki, S.O.; Kanba, S.; Kiyohara, Y.; et al. Insulin resistance is associated with the pathology of Alzheimer disease: The Hisayama study. Neurology 2010, 75, 764–770.
- Bedse, G.; Di Domenico, F.; Serviddio, G.; Cassano, T. Aberrant insulin signaling in Alzheimer’s disease: Current knowledge. Front. Neurosci. 2015, 9, 204.
- Abolhassani, N.; Leon, J.; Sheng, Z.; Oka, S.; Hamasaki, H.; Iwaki, T.; Nakabeppu, Y. Molecular pathophysiology of impaired glucose metabolism, mitochondrial dysfunction, and oxidative DNA damage in Alzheimer’s disease brain. Mech. Ageing Dev. 2017, 161, 95–104.
- Cortes, N.; Andrade, V.; Maccioni, R.B. Behavioral and Neuropsychiatric Disorders in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 63, 899–910.
- Donovan, N.J.; Locascio, J.J.; Marshall, G.A.; Gatchel, J.; Hanseeuw, B.J.; Rentz, D.M.; Johnson, K.A.; Sperling, R.A.; Harvard Aging Brain, S. Longitudinal Association of Amyloid Beta and Anxious-Depressive Symptoms in Cognitively Normal Older Adults. Am. J. Psychiatry 2018, 175, 530–537.
- Ownby, R.L.; Crocco, E.; Acevedo, A.; John, V.; Loewenstein, D. Depression and risk for Alzheimer disease: Systematic review, meta-analysis, and metaregression analysis. Arch. Gen. Psychiatry 2006, 63, 530–538.
- Green, R.C.; Cupples, L.A.; Kurz, A.; Auerbach, S.; Go, R.; Sadovnick, D.; Duara, R.; Kukull, W.A.; Chui, H.; Edeki, T.; et al. Depression as a risk factor for Alzheimer disease: The MIRAGE Study. Arch. Neurol. 2003, 60, 753–759.
- Royall, D.R.; Palmer, R.F. Alzheimer’s disease pathology does not mediate the association between depressive symptoms and subsequent cognitive decline. Alzheimers Dement. 2013, 9, 318–325.
- Chistyakov, D.V.; Astakhova, A.A.; Sergeeva, M.G. Resolution of inflammation and mood disorders. Exp. Mol. Pathol. 2018, 105, 190–201.
- Galts, C.P.C.; Bettio, L.E.B.; Jewett, D.C.; Yang, C.C.; Brocardo, P.S.; Rodrigues, A.L.S.; Thacker, J.S.; Gil-Mohapel, J. Depression in neurodegenerative diseases: Common mechanisms and current treatment options. Neurosci. Biobehav. Rev. 2019, 102, 56–84.
- Rojo, L.E.; Fernandez, J.A.; Maccioni, A.A.; Jimenez, J.M.; Maccioni, R.B. Neuroinflammation: Implications for the pathogenesis and molecular diagnosis of Alzheimer’s disease. Arch. Med. Res. 2008, 39, 1–16.
- Maccioni, R.B.; Navarrete, L.P.; Gonzalez, A.; Gonzalez-Canacer, A.; Guzman-Martinez, L.; Cortes, N. Inflammation: A Major Target for Compounds to Control Alzheimer’s Disease. J. Alzheimers Dis. 2020, 76, 1199–1213.
- Canet, G.; Hernandez, C.; Zussy, C.; Chevallier, N.; Desrumaux, C.; Givalois, L. Is AD a Stress-Related Disorder? Focus on the HPA Axis and Its Promising Therapeutic Targets. Front. Aging Neurosci. 2019, 11, 269.
- Morales, I.; Guzman-Martinez, L.; Cerda-Troncoso, C.; Farias, G.A.; Maccioni, R.B. Neuroinflammation in the pathogenesis of Alzheimer’s disease. A rational framework for the search of novel therapeutic approaches. Front. Cell Neurosci. 2014, 8, 112.
- Tanzi, R.E.; McClatchey, A.I.; Lamperti, E.D.; Villa-Komaroff, L.; Gusella, J.F.; Neve, R.L. Protease inhibitor domain encoded by an amyloid protein precursor mRNA associated with Alzheimer’s disease. Nature 1988, 331, 528–530.
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917.
- Muralidar, S.; Ambi, S.V.; Sekaran, S.; Thirumalai, D.; Palaniappan, B. Role of tau protein in Alzheimer’s disease: The prime pathological player. Int. J. Biol. Macromol. 2020, 163, 1599–1617.
- Shamsi, A.; Al Shahwan, M.; Ahamad, S.; Hassan, M.I.; Ahmad, F.; Islam, A. Spectroscopic, calorimetric and molecular docking insight into the interaction of Alzheimer’s drug donepezil with human transferrin: Implications of Alzheimer’s drug. J. Biomol. Struct. Dyn. 2020, 38, 1094–1102.
- Fernandez, J.A.; Rojo, L.; Kuljis, R.O.; Maccioni, R.B. The damage signals hypothesis of Alzheimer’s disease pathogenesis. J. Alzheimers Dis. 2008, 14, 329–333.
- Maccioni, R.B.; Rojo, L.E.; Fernandez, J.A.; Kuljis, R.O. The role of neuroimmunomodulation in Alzheimer’s disease. Ann. NY Acad. Sci. 2009, 1153, 240–246.
- Liu, M.; Dexheimer, T.; Sui, D.; Hovde, S.; Deng, X.; Kwok, R.; Bochar, D.A.; Kuo, M.H. Hyperphosphorylated tau aggregation and cytotoxicity modulators screen identified prescription drugs linked to Alzheimer’s disease and cognitive functions. Sci. Rep. 2020, 10, 1–14.
- Neve, R.L.; Harris, P.; Kosik, K.S.; Kurnit, D.M.; Donlon, T.A. Identification of cDNA clones for the human microtubule-associated protein tau and chromosomal localization of the genes for tau and microtubule-associated protein 2. Brain Res. 1986, 387, 271–280.
- Schweers, O.; Schonbrunn-Hanebeck, E.; Marx, A.; Mandelkow, E. Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for beta-structure. J. Biol. Chem. 1994, 269, 24290–24297.
- Luna-Viramontes, N.I.; Campa-Córdoba, B.B.; Ontiveros-Torres, M.Á.; Harrington, C.R.; Villanueva-Fierro, I.; Guadarrama-Ortíz, P.; Garcés-Ramírez, L.; de la Cruz, F.; Hernandes-Alejandro, M.; Martínez-Robles, S.; et al. PHF-Core Tau as the Potential Initiating Event for Tau Pathology in Alzheimer’s Disease. Front. Cell Neurosci. 2020, 14, 247.
- Gong, C.X.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K. Post-translational modifications of tau protein in Alzheimer’s disease. J. Neural. Transm. Vienna 2005, 112, 813–838.
- Maccioni, R.B.; Otth, C.; Concha, I.I.; Munoz, J.P. The protein kinase Cdk5. Structural aspects, roles in neurogenesis and involvement in Alzheimer’s pathology. Eur. J. Biochem. 2001, 268, 1518–1527.
- Martin, L.; Latypova, X.; Terro, F. Post-translational modifications of tau protein: Implications for Alzheimer’s disease. Neurochem. Int. 2011, 58, 458–471.
- Alonso, A.C.; Grundke-Iqbal, I.; Iqbal, K. Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat. Med. 1996, 2, 783–787.
- Gendron, T.F.; Petrucelli, L. The role of tau in neurodegeneration. Mol. Neurodegener. 2009, 4, 1–19.
- Gotz, J.; Halliday, G.; Nisbet, R.M. Molecular Pathogenesis of the Tauopathies. Annu. Rev. Pathol. 2019, 14, 239–261.
- Bennecib, M.; Gong, C.X.; Grundke-Iqbal, I.; Iqbal, K. Role of protein phosphatase-2A and -1 in the regulation of GSK-3, cdk5 and cdc2 and the phosphorylation of tau in rat forebrain. FEBS Lett. 2000, 485, 87–93.
- Sun, K.H.; de Pablo, Y.; Vincent, F.; Shah, K. Deregulated Cdk5 promotes oxidative stress and mitochondrial dysfunction. J. Neurochem. 2008, 107, 265–278.
- Chung, S.H. Aberrant phosphorylation in the pathogenesis of Alzheimer’s disease. BMB Rep. 2009, 42, 467–474.
- Dhavan, R.; Tsai, L.H. A decade of CDK5. Nat. Rev. Mol. Cell Biol. 2001, 2, 749–759.
- Zambrano, C.A.; Egana, J.T.; Nunez, M.T.; Maccioni, R.B.; Gonzalez-Billault, C. Oxidative stress promotes tau dephosphorylation in neuronal cells: The roles of cdk5 and PP1. Free Radic. Biol. Med. 2004, 36, 1393–1402.
- Takahashi, M.; Tsujioka, Y.; Yamada, T.; Tsuboi, Y.; Okada, H.; Yamamoto, T.; Liposits, Z. Glycosylation of microtubule-associated protein tau in Alzheimer’s disease brain. Acta. Neuropathol. 1999, 97, 635–641.
- Liu, F.; Zaidi, T.; Iqbal, K.; Grundke-Iqbal, I.; Merkle, R.K.; Gong, C.X. Role of glycosylation in hyperphosphorylation of tau in Alzheimer’s disease. FEBS Lett. 2002, 512, 101–106.
- Planel, E.; Yasutake, K.; Fujita, S.C.; Ishiguro, K. Inhibition of protein phosphatase 2A overrides tau protein kinase I/glycogen synthase kinase 3 beta and cyclin-dependent kinase 5 inhibition and results in tau hyperphosphorylation in the hippocampus of starved mouse. J. Biol. Chem. 2001, 276, 34298–34306.
- Shamsi, A.; Anwar, S.; Mohammad, T.; Alajmi, M.F.; Hussain, A.; Rehman, M.T.; Hasan, G.M.; Islam, A.; Hassan, M.I. MARK4 Inhibited by AChE Inhibitors, Donepezil and Rivastigmine Tartrate: Insights into Alzheimer’s Disease Therapy. Biomolecules 2020, 10, 789.
- Waseem, R.; Anwar, S.; Khan, S.; Shamsi, A.; Hassan, M.I.; Anjum, F.; Shafie, A.; Islam, A.; Yadav, D.K. MAP/Microtubule Affinity Regulating Kinase 4 Inhibitory Potential of Irisin: A New Therapeutic Strategy to Combat Cancer and Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 10986.
- Saint-Aubert, L.; Lemoine, L.; Chiotis, K.; Leuzy, A.; Rodriguez-Vieitez, E.; Nordberg, A. Tau PET imaging: Present and future directions. Mol. Neurodegener. 2017, 12, 1–21.
- Shi, Y.; Murzin, A.G.; Falcon, B.; Epstein, A.; Machin, J.; Tempest, P.; Newell, K.L.; Vidal, R.; Garringer, H.J.; Sahara, N.; et al. Cryo-EM structures of tau filaments from Alzheimer’s disease with PET ligand APN-1607. Acta. Neuropathol. 2021, 141, 697–708.
- Cortes, N.; Guzman-Martinez, L.; Andrade, V.; Gonzalez, A.; Maccioni, R.B. CDK5: A Unique CDK and Its Multiple Roles in the Nervous System. J. Alzheimers Dis. 2019, 68, 843–855.
- Braak, H.; Braak, E. Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathol. 1991, 1, 213–216.
- Mohamed, N.V.; Herrou, T.; Plouffe, V.; Piperno, N.; Leclerc, N. Spreading of tau pathology in Alzheimer’s disease by cell-to-cell transmission. Eur. J. Neurosci. 2013, 37, 1939–1948.
More
Information
Subjects:
Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Entry Collection:
Neurodegeneration
Revisions:
2 times
(View History)
Update Date:
28 Apr 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No