 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Carlos P. Dopazo | -- | 3236 | 2022-04-27 16:44:38 | | | |
| 2 | Peter Tang | -9 word(s) | 3227 | 2022-04-28 04:07:01 | | |
Video Upload Options
Infectious pancreatic necrosis (IPN) is a disease of great concern in aquaculture, mainly among salmonid farmers, since losses in salmonid fish—mostly very young rainbow trout (Salmo gairdnery) fry and Atlantic salmon (Salmo salar) post-smolt—frequently reach 80–90% of stocks. The virus causing the typical signs of the IPN disease in salmonids, named infectious pancreatic necrosis virus (IPNV), has also been isolated from other fish species either suffering related diseases (then named IPNV-like virus) or asymptomatic; the general term aquabirnavirus is used to encompass all these viruses. Aquabirnaviruses are non-enveloped, icosahedral bisegmented dsRNA viruses, whose genome codifies five viral proteins, three of which are structural, and one of them is an RNA-dependent RNA polymerase.
1. The Structure and General Characteristics
|
Stp 1 |
Gtp 2 |
Type Strain |
Geogr Origin 3 |
|
|---|---|---|---|---|
|
A |
A1 |
1 |
WB |
USA |
|
A2 |
5 |
Sp |
Denmark |
|
|
A3 |
2 |
Ab |
Denmark |
|
|
A4 |
6 |
He |
Germany |
|
|
A5 |
3 |
Te |
UK |
|
|
A6 |
3 |
C1 |
Canada |
|
|
A7 |
4 |
C2 |
Canada |
|
|
A8 |
4 |
C3 |
Canada |
|
|
A9 |
1 |
Ja |
Canada |
|
|
B |
B1 |
– |
TV-1 |
UK |
|
7 |
MaBV 4 |
Japan |
||
1—Serogroup/Serotype; 2—Genotype; 3—Geographic origin; 4—Marine birnavirus.
2. The Viral Genome
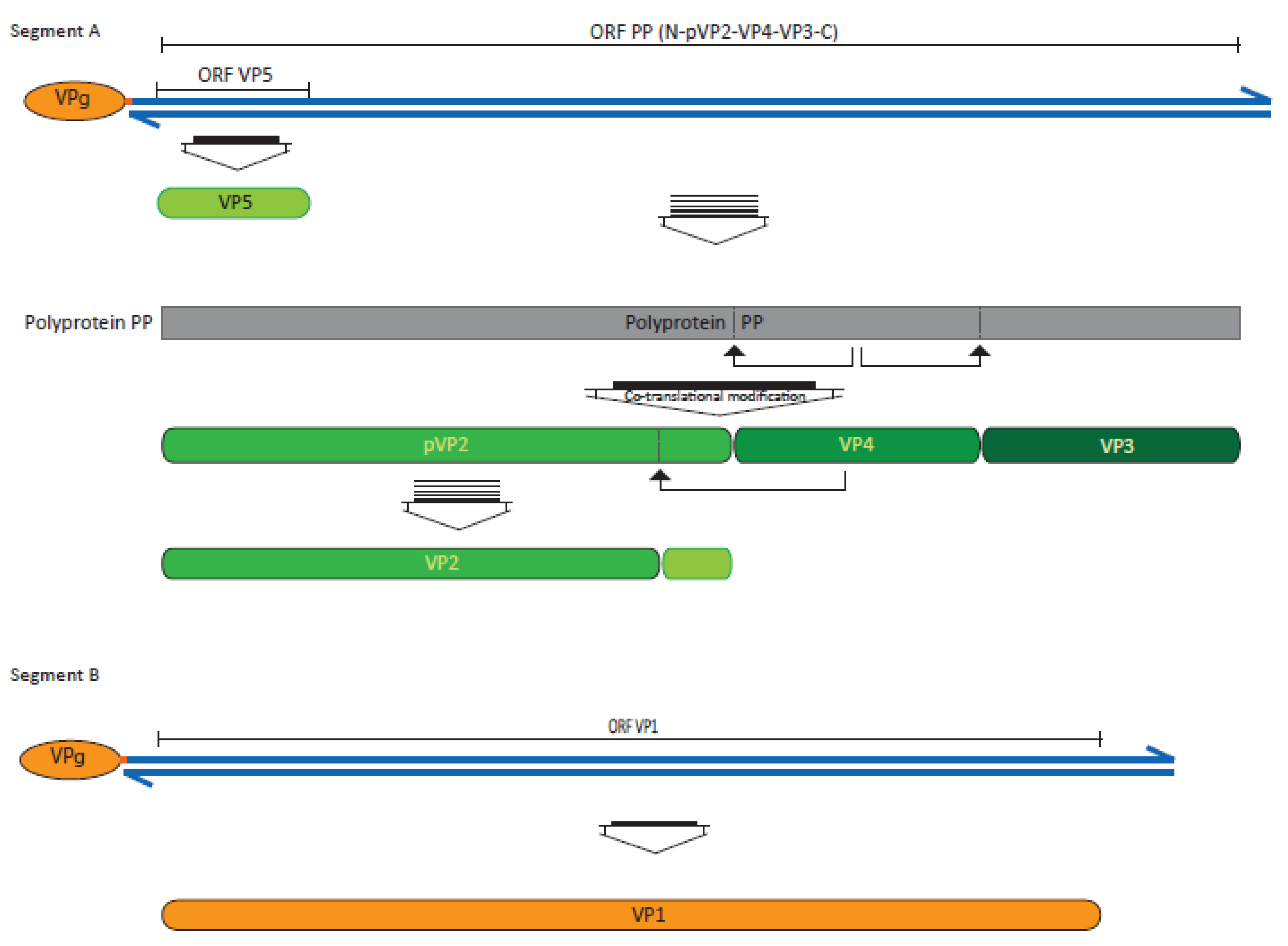
3. The Viral Proteins and Their Function
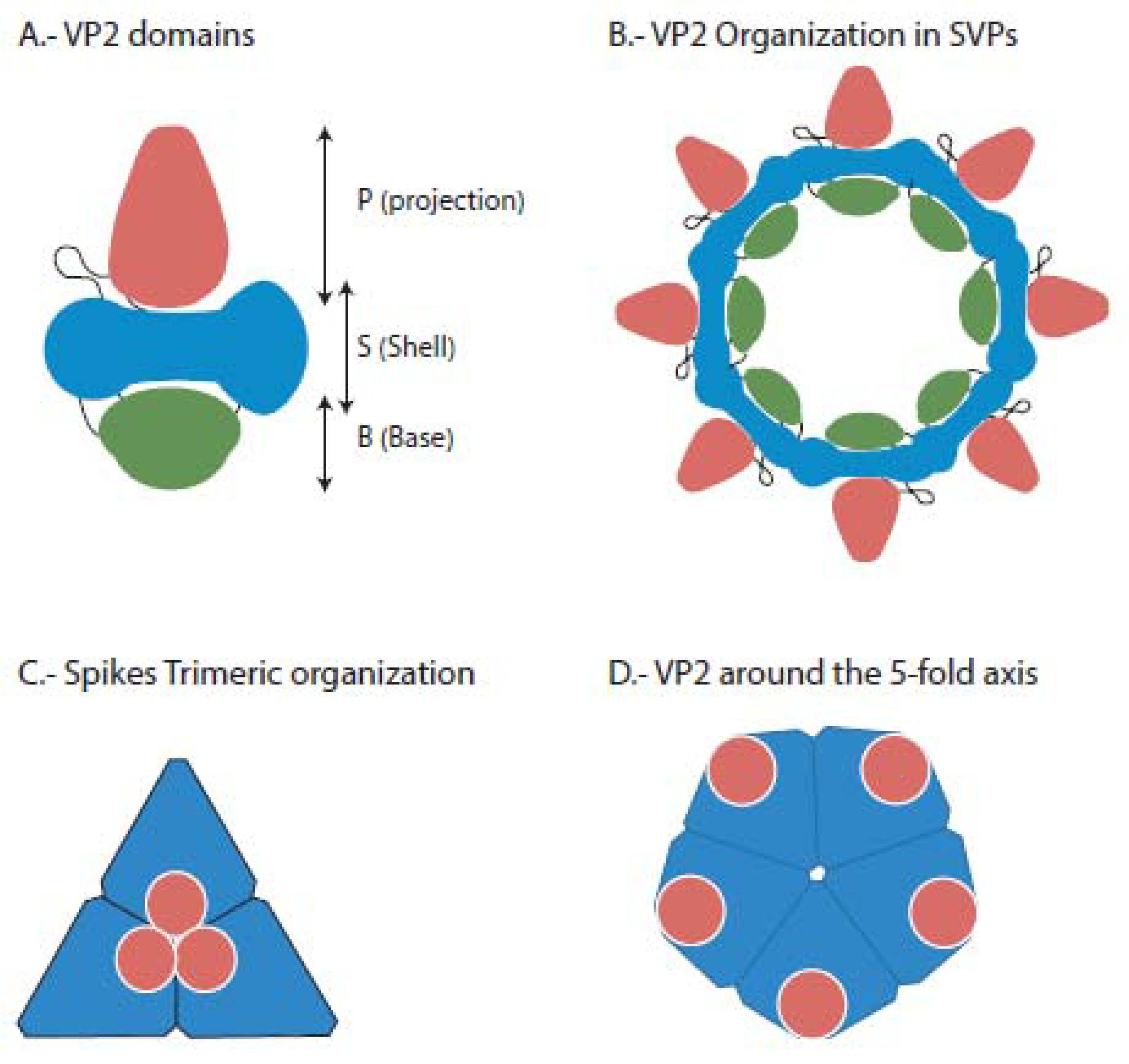

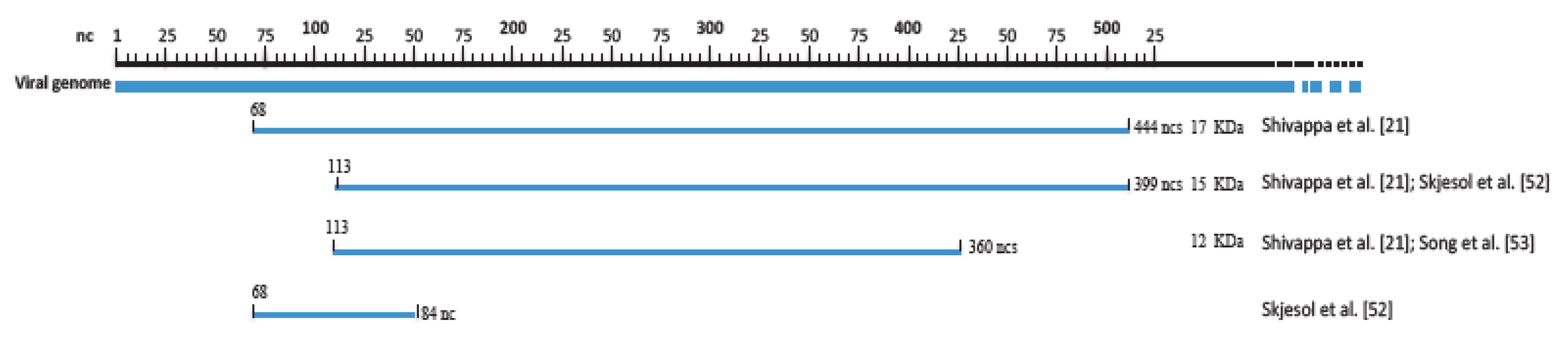
4. Replication Cycle
5. IPNV and Persistence
References
- Delmas, D.; Attoui, H.; Ghosh, S.; Malik, Y.S.; Mundt, E.; Vakharia, V.N. ICTV Report Consortium, ICTV Virus Taxonomy Profile: Birnaviridae. J. Gen. Virol. 2019, 100, 5–6.
- Munro, E.S.; Midtlyng, P.J. Infectious pancreatic necrosis virus and associated aquatic birnavirus. In Fish Diseases and Disorders Vol. 3: Viral, Bacterial and Fungal Infections, 2nd ed.; Woo, P.T.K., Bruno, D.W., Eds.; CAB International: Cambridge, MA, USA, 2011; pp. 1–65.
- Dobos, P.; Roberts, T.E. The molecular biology of infectious pancreatic necrosis virus: A review. Can. J. Microbiol. 1983, 29, 377–384.
- Lago, L.; Rodríguez, J.F.; Bandín, I.; Dopazo, C.P. Aquabirnavirus polyploidy: A new strategy to modulate virulence? J. Gen. Virol. 2016, 97, 1168–1177.
- Wolf, K. Fish Viruses and Fish Viral Diseases; Comstock Pbl Ass., Cornwell Univ. Press: New York, NY, USA, 1988; p. 576.
- Reno, P.W. Infectious pancreatic necrosis virus and associated aquatic birnavirus. In Fish Diseases and Disorders, Vol. 3: Viral, Bacterial and Fungal Infections; Woo, P.T.K., Bruno, D.W., Eds.; CAB International: New York, NY, USA, 1999; pp. 1–55.
- Hill, B.J.; Way, K. Serological classification of infectious pancreatic necrosis (IPN) virus and other aquatic birnaviruses. Ann. Rev. Fish Dis. 1995, 5, 55–77.
- Blake, S.L.; Ma, J.-Y.; Caporale, D.A.; Jairath, S.; Nicholson, B.L. Phylogenetic relationships of aquatic birnaviruses based on deduced amino acid sequences of genome segment A cDNA. Dis. Aquat. Org. 2001, 45, 89–102.
- Nishizawa, T.; Kinoshita, S.; Yoshimizu, M. An approach for genogrouping of Japanese isolates of aquabirnaviruses in a new genogroup, VII, based on the VP2/NS junction region. J. Gen. Virol. 2005, 86, 1973–1978.
- Galloux, M.; Chevalier, C.; Henrry, C.; Huet, J.C.; Costa, B.D.; Delmas, B. Peptides resulting from the pVP2 C-terminal processing are present in infectious pancreatic necrosis virus particles. J. Gen. Virol. 2004, 85, 2231–2236.
- Dobos, P. Protein-primed RNA synthesis in vitro by the virion-associated RNA polymerase of infectious pancreatic necrosis virus. Virology 1995, 208, 19–25.
- Magyar, G.; Chung, H.K.; Dobos, P. Conversion of VP1 to VPg in cells infected with infectious pancreatic necrosis virus. Virology 1988, 245, 142–150.
- Weber, S.; Fichner, D.; Mettenleiter, T.C.; Mundt, E. Expression of VP5 of infectious pancreatic necrosis virus strain VR299 is initiated at the second in-frame start codon. J. Gen. Virol. 2001, 82, 805–812.
- Boot, H.; Pritz-Verschuren, B.E.P. Modifications of the 3′UTR stem-loop of infectious bursal disease virus are allowed without influencing replication or virulence. Nucl. Acids Res. 2004, 32, 211–222.
- Rivas-Aravena, A.; Muñoz, P.; Jorquera, P.; Diaz, A.; Reinoso, C.; González-Catrilelbún, S.; Sandino, A.M. Study of RNA-A Initiation Translation of The Infectious Pancreatic Necrosis Virus. Virus Res. 2017, 240, 121–129.
- Shivappa, R.; Song, H.; Yao, K.; Aas-Eng, A.; Evensen, Ø.; Vakharia, V.N. Molecular characterization of Sp serotype strains of infectious pancreatic necrosis virus exhibiting differences in virulence. Dis. Aquat. Org. 2004, 61, 23–32.
- Gondenberg, N.M.; Steinberg, B.E. Surface charge: A key determinant of protein localization and function. Cancer Res. 2010, 70, 1277–1280.
- Villanueva, R.A.; Guacucano, M.; Pizarro, J.; Sandino, A.M. Inhibition of virion-associated IPNV polymerase, VP1, by radiolabeled nucleotide analogs. Virus Res. 2005, 112, 132–135.
- Dobos, P. In vitro guanylation of infectious pancreatic necrosis virus polypeptide VP1. Virology 1993, 193, 403–413.
- Xu, H.-T.; Si, W.D.; Dobos, P. Mapping the site of guanylylation on VP1, the protein primer for infectious pancreatic necrosis virus RNA synthesis. Virology 2004, 322, 199–210.
- Coulibaly, F.; Chevalier, C.; Gutsche, I.; Pous, J.; Navaza, J.; Bressanelli, S.; Delmas, B.; Rey, F.A. The birnavirus crystal structure reveals structural relationships among icosahedral viruses. Cell 2005, 120, 761–772.
- Coulibaly, F.; Chevalier, C.; Delmas, B.; Rey, F.A. Crystal Structure of an Aquabirnavirus Particle: Insights into Antigenic Diversity and Virulence Determinism. J. Virol. 2010, 84, 1792–1799.
- Caswell-Reno, P.; Reno, P.W.; Nicholson, B. Monoclonal antibodies to infectious pancreatic necrosis virus: Analysis of viral epitopes and comparison of different isolates. J. Gen. Virol. 1986, 67, 2193–2205.
- Azad, A.A.; Jagadish, M.N.; Brown, M.A.; Hudson, P.J. Deletion mapping and expression in Escherichia coli of the large genomic segment of birnavirus. Virology 1987, 161, 145–152.
- Håvarstein, L.S.; Kalland, K.H.; Christie, K.E.; Endresen, C. Sequence of the large double-stranded RNA segment of the NI strain of infectious pancreatic necrosis virus: A comparison with other Birnaviridae. J. Gen. Virol. 1990, 71, 299–308.
- Heppell, J.L.; Tarrab, E.; Lecomte, J.; Berthiaume, L.; Arella, M. Strain variability and localization of important epitopes on the major structural protein (VP2) of infectious pancreatic necrosis virus. Virology 1995, 214, 40–49.
- Dobos, P. The molecular biology of infectious pancreatic necrosis virus (IPNV). Ann. Rev. Fish. Dis. 1995, 5, 25–54.
- Frost, P.; Håvarstein, L.S.; Lygren, B.; Stahl, S.; Endresen, C.; Christie, K.E. Mapping of neutralization epitopes on infectious pancreatic necrosis virus. J. Gen. Virol. 1995, 76, 1165–1172.
- Darragh, E.A.; MacDonald, R.D. A host range restriction in infectious pancreatic necrosis virus maps to the large RNA segment and involves virus attachment to the cell surface. Virology 1982, 123, 264–272.
- Delgui, L.; Ona, A.; Gutierrez, S.; Luque, D.; Navarro, A.; Caston, J.R.; Rodríguez, J.F. The capsid protein of infectious bursal disease virus contains functional alpha 4 beta 1 integrin ligand motif. Virology 2009, 386, 360–372.
- Hjalmarsson, A.; Everitt, E. Identification of IPNV- specified components released from productively infected RTG-2 cell following massive cytopathic effect. Arch. Virol. 1999, 144, 1487–1501.
- Espinoza, J.C.; Hjalmarsson, A.; Everitt, E.; Kuznar, J. Temporal and subcellular localization of infectious pancreatic necrosis virus structural proteins. Arch. Virol. 2000, 145, 739–748.
- Fridholm, H.; Everitt, E. Virion glycosylation governs integrity and infectivity of infectious pancreatic necrosis virus. J. Fish Dis. 2011, 34, 663–675.
- Ferrero, D.; Garriga, D.; Navarro, A.; Rodríguez, J.F.; Verdaguer, N. Infectious Bursal Disease Virus VP3 Upregulates VP1-Mediated RNA- Dependent RNA Replication. J. Virol. 2015, 89, 11165–11168.
- Chiu, C.-L.; Wu, J.-L.; Her, G.-M.; Chou, Y.-L.; Hong, J.-R. Aquatic birnavirus capsid protein, VP3, induces apoptosis via the Bad-mediated mitochondria pathway in fish and mouse cells. Apoptosis 2010, 15, 653–668.
- Boot, H.J.; ter Huurne, A.A.H.M.; Hoekman, A.J.W.; Pol, J.M.; Gielkens, A.L.J.; Peeters, B.P.H. Exchange of the C-Terminal Part of VP3 from Very Virulent Infectious Bursal Disease Virus Results in an Attenuated Virus with a Unique Antigenic Structure. J. Virol. 2002, 76, 10346–10355.
- Casañas, A.; Navarro, A.; Ferrer-Orta, C.; González, D.; Rodríguez, J.F.; Verdaguer, N. tructural Insights into the Multifunctional Protein VP3 of Birnaviruses. Structure 2008, 16, 29–37.
- Nicholson, B.L. Use of monoclonal antibodies in identification and characterization of fish viruses. Ann. Rev. Fish Dis. 1993, 3, 241–257.
- Pedersen, T.; Skjesol, A.; Jørgensen, J.B. VP3, a Structural Protein of Infectious Pancreatic Necrosis Virus, Interacts with RNA-Dependent RNA Polymerase VP1 and with Double-Stranded RNA. J. Virol. 2007, 81, 6652–6663.
- Tacken, M.G.; Peeters, B.P.; Thomas, A.A.; Rottier, P.J.; Boot, H.J. Infectious bursal disease virus capsid protein VP3 interacts both with VP1, the RNA-dependent RNA polymerase, and with viral double-stranded RNA. J. Virol. 2002, 76, 11301–11311.
- Bahar, M.W.; Sarin, L.P.; Graham, S.C.; Pang, J.; Bamford, D.H.; Stuart, D.I.; Grimes, J.M. Structure of a VP1-VP3 complex suggests how birnaviruses package the VP1 polymerase. J. Virol. 2013, 87, 3229–3236.
- Birghan, C.; Mundt, E.; Gorbalenya, A.E. A non-canonical lon proteinase lacking the ATPase domain employs the ser-Lys catalytic dyad to exercise broad control over the life cycle of a double-stranded RNA virus. EMBO J. 2000, 19, 114–123.
- Lauksund, S.; Greiner-Tollersrud, L.; Chang, C.-J.; Robertsen, B. Infectious pancreatic necrosis virus proteins VP2, VP3, VP4 and VP5 antagonize IFNa1 promoter activation while VP1 induces IFNa1. Virus Res. 2015, 196, 113–121.
- Magyar, G.; Dobos, P. Expression of infectious pancreatic necrosis virus polyprotein and vp1 in insect cells and the detection of the polyprotein in purified virus. Virology 1994, 198, 437–445.
- Mundt, E.; Beyer, J.; Muller, H. Identification of a novel viral protein in infectious bursal disease virus-infected cells. J. Gen. Virol. 1995, 76, 437–443.
- Heppell, J.; Tarrab, T.; Berthiaume, L.; Leeomte, J.; Arella, M. Characterization of the small open reading frame on genome segment A of infectious pancreatic necrosis virus. J. Gen. Virol. 1995, 76, 2091–2096.
- Skjesol, A.; Skjæveland, I.; Elnæs, M.; Timmerhaus, G.; Fredriksen, B.N.; Jørgensen, S.N.; Krasnov, A.; Jørgensen, J.B. IPNV with high and low virulence: Host immune responses and viral mutations during infection. Virol. J. 2011, 8, 396.
- Song, H.; Baxter-Roshek, J.L.; Dinman, J.D.; Vakharia, V.N. Efficient expression of the 15-kDa form of infectious pancreatic necrosis virus VP5 by suppression of a UGA codon. Virus Res. 2006, 122, 61–68.
- Santi, N.; Song, H.; Vakharia, V.N.; Evensen, Ø. Infectious pancreatic necrosis virus VP5 is dispensable for virulence and persistence. J. Virol. 2005, 79, 9206–9216.
- Hong, J.R.; Gong, H.Y.; Wu, J.L. IPNV VP5, a novel anti-apoptosis gene of the Bcl-2 family, regulates Mcl-1 and viral protein expression. Virology 2002, 295, 217–229.
- Liu, M.; Vakharia, V.N. Nonstructural Protein of Infectious Bursal Disease Virus Inhibits Apoptosis at the Early Stage of Virus Infection. J. Virol. 2006, 80, 3369–3377.
- Ortega, C.; Rodríguez, S.; Espinoza, J.C.; Kuznar, J.; Romero, A.; Enríquez, R. Relationship between apoptosis and the BH2 domain sequence of the VP5 peptide of infectious pancreatic necrosis virus. Rev. Mvz. Córdoba 2014, 19, 3990–4002.
- Hong, J.R.; Lin, T.L.; Hsu, Y.L.; Wu, J.L. Apoptosis precedes necrosis of fish cell line by infectious pancreatic necrosis virus. Virology 1998, 250, 76–84.
- Ulrich, K.; Wehner, S.; Bekaert, M.; Di Paola, N.; Dilcher, M.; Muir, K.F.; Taggart, J.B.; Matejusova, I.; Weidmann, M. Molecular epidemiological study on Infectious Pancreatic Necrosis Virus isolates from aquafarms in Scotland over three decades. J. Gen. Virol. 2018, 99, 1567–1581.
- Skjesol, A.; Aamo, T.; Hegseth, M.N.; Robertsen, B.; Jørgensen, J.B. The interplay between infectious pancreatic necrosis virus (IPNV) and the IFN system: IFN signaling is inhibited by IPNV infection. Virus Res. 2009, 143, 53–60.
- Nouën, C.L.; Toquin, D.; Müller, H.; Raue, R.; Kean, K.M.; Langlois, P.; Cherbonnel, M.; Eterradossi, N. Different Domains of the RNA Polymerase of Infectious Bursal Disease Virus Contribute to Virulence. PLoS ONE 2012, 7, e28064.
- Ortega, C.; Enríquez, R. Factors associated with cellular infection by the infectious pancreatic necrosis virus (IPNV). Arch. Med. Vet. 2007, 39, 7–18.
- Kuznar, J.; Soler, M.; Farias, G.; Espinoza, J.C. Attachment and entry of infectious pancreatic necrosis virus (IPNV) into CHSE-214 cells. Arch. Virol. 1995, 140, 1833–1840.
- Martin, M.C.S.; Villanueva, R.A.; Jashes, M.; Sandino, A.M. Molecular characterization of IPNV RNA replication intermediates during the viral infective cycle. Virus Res. 2009, 144, 344–349.
- Villanueva, R.A.; Galaz, J.L.; Valdés, J.A.; Jashés, M.M.; Sandino, A.M. Genome assembly and particle maturation of the birnavirus infectious pancreatic necrosis virus. J. Virol. 2004, 78, 13829–13838.
- Lombardo, E.; Maraver, A.; Espinosa, I.; Fernandez-Arias, A.; Rodriguez, J.F. VP5, the nonstructural polypeptide of infectious bursal disease virus, accumulates within the host plasma membrane and induces cell lysis. Virology 2000, 277, 345–357.
- Wu, Y.; Hong, L.; Ye, J.; Huang, Z.; Zhou, J. The VP5 protein of infectious bursal disease virus promotes virion release from infected cells and is not involved in cell death. Arch. Virol. 2009, 154, 1873–1882.
- Marjara, I.S.; Thu, B.J.; Evensen, Ø. Differentially expressed genes following persistent infection with infectious pancreatic necrosis virus in vitro and in vivo. Fish Shellfih Immunol. 2010, 28, 845–853.
- Julin, K.; Johansen, L.H.; Sommer, A.I.; Jørgense, J.B. Persistent infections with infectious pancreatic necrosis virus (IPNV) of different virulence in Atlantic salmon, Salmo salar L. J. Fish Dis. 2015, 38, 1005–1019.


