Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sarfaraz K. Niazi | -- | 1814 | 2022-04-27 03:09:53 | | | |
| 2 | Conner Chen | Meta information modification | 1814 | 2022-04-27 05:21:07 | | | | |
| 3 | Conner Chen | Meta information modification | 1814 | 2022-04-27 05:21:51 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Niazi, S. Analytical Assessment of Biosimilars. Encyclopedia. Available online: https://encyclopedia.pub/entry/22330 (accessed on 01 August 2026).
Niazi S. Analytical Assessment of Biosimilars. Encyclopedia. Available at: https://encyclopedia.pub/entry/22330. Accessed August 01, 2026.
Niazi, Sarfaraz. "Analytical Assessment of Biosimilars" Encyclopedia, https://encyclopedia.pub/entry/22330 (accessed August 01, 2026).
Niazi, S. (2022, April 27). Analytical Assessment of Biosimilars. In Encyclopedia. https://encyclopedia.pub/entry/22330
Niazi, Sarfaraz. "Analytical Assessment of Biosimilars." Encyclopedia. Web. 27 April, 2022.
Copy Citation
The analytical assessment includes testing physicochemical and functional attributes to establish a claim of biosimilarity. How closely a biosimilar candidate should match the reference product will remain questionable since a reference product is approved based on whatever quality attributes it presents; a biosimilar candidate, on the other hand, must match these quality attributes, even if the reference product’s attributes are not the most desirable.
biosimilars
FDA
EMA
SIMILAR BIOLOGICS
SIMILAR BIOTHERAPEUTICS
1. Analytical Assessment
The analytical assessment includes testing physicochemical and functional attributes to establish a claim of biosimilarity. How closely a biosimilar candidate should match the reference product will remain questionable since a reference product is approved based on whatever quality attributes it presents; a biosimilar candidate, on the other hand, must match these quality attributes, even if the reference product’s attributes are not the most desirable. An earlier FDA guideline, “Statistical Approaches to Evaluate Analytical Similarity” [1], recommended a rigorous statistical approach for establishing similarity that turned out to be overkill, and the guidance was withdrawn [2] and replaced with a new guideline [3] in response to the author’s citizen petition [4]. The new guideline changed the terminology from “analytical testing” to “analytical assessment”, meaning an overall evaluation rather than specific test results. For example, this eliminated the controversial tier 1 assessment of quality attributes. In addition, this required setting up arbitrary equivalence criteria such as 1.5 × SD of the reference product to define the 90% confidence limit of the biosimilar candidate, with no justification for the factor of 1.5 used. Instead, the new guideline suggests using a range approach that is more practical and scientifically sound. However, as all biosimilar products approved by the FDA followed the earlier guideline, there is a lot of analytical testing that would be avoidable in the future. For example, companies have submitted different number of studies for adalimumab—25 by Pfizer and 71 by Boehringer—to achieve the same goal [5].
The EMA provides more comprehensive guidance divided into immunogenicity testing, quality issues, clinical and non-clinical testing, pharmacokinetic modeling, and guidance on changing the manufacturing process of recombinant drugs [6]. In addition, the product-specific guidelines of the EMA are of great value for biosimilar developers [7].
Most regulatory guidelines suggest that a biosimilar candidate’s quality target product profile (QTPP) should be based on the data collected on the chosen reference product, including publicly available information and data obtained from the extensive characterization of the reference medicinal product [8]. The QTPPs are well defined, and there may not be any need to establish their relative importance and assign a criticality factor to plan the testing, as these are now well-established. However, as suggested below, the developers should challenge the merits of testing an attribute. Quality attributes fall into two categories, product- or process-related.
2. Product-Related Attributes
Product-related attributes (not to be confused with the drug product that is the finished form) relate to the production of proteins by cells that can make exact copies of the protein [9] (Figure 1). Still, after the protein is made, other variations (e.g., add-ons and changes) may occur, such as adding sugar molecules or modifying certain amino acids. The expression system determines the product-related attributes with as little manipulation as possible. The QTPP profile must match the reference product and undergo the well-established testing required. Tests of the biosimilar must be conducted side-by-side with tests of the reference product to remove any test method variability, as the test methods need not be validated (Figure 1).
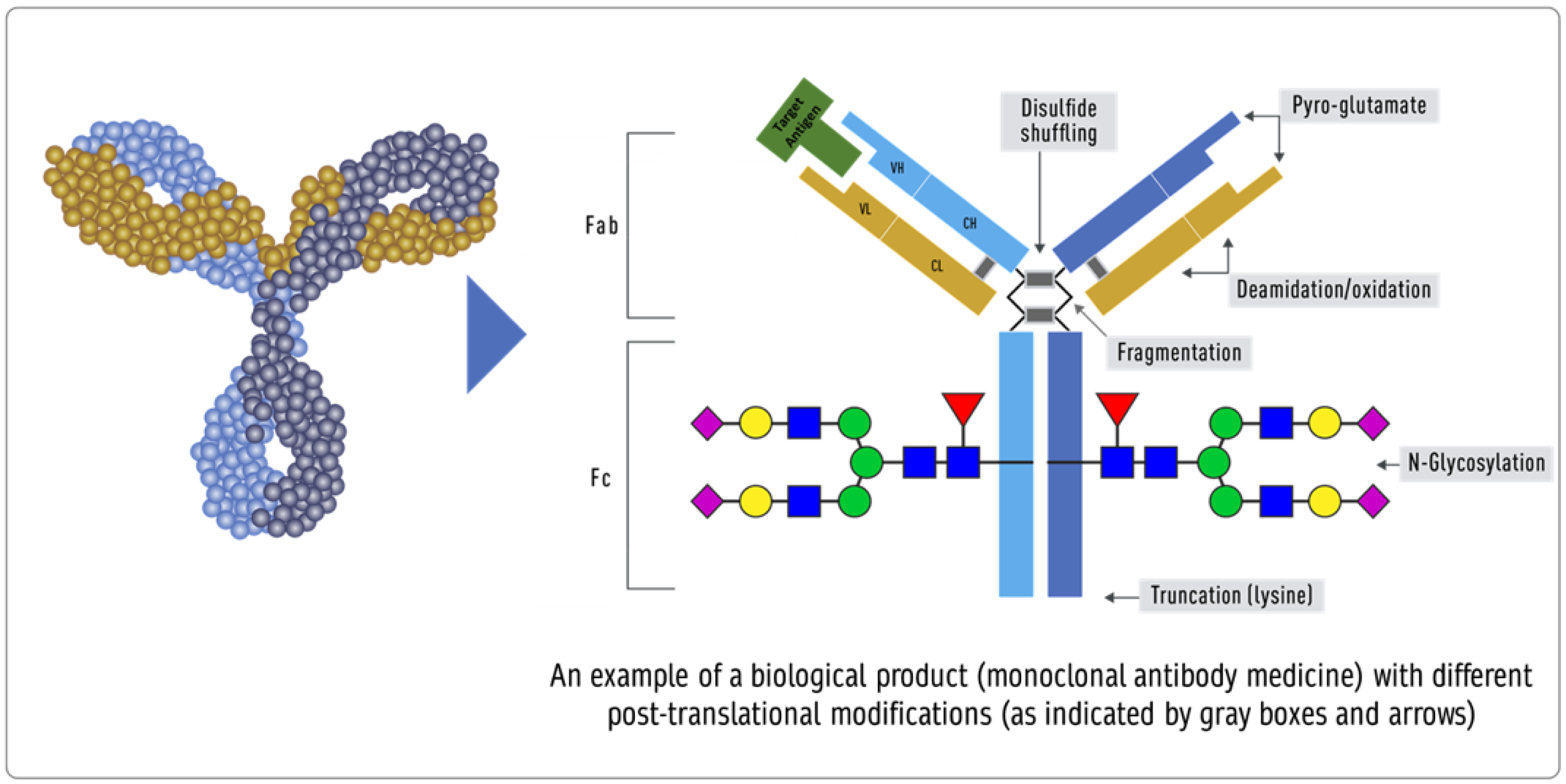
Figure 1. Inherent variations in biological products (FDA: https://www.fda.gov/drugs/biosimilars/curriculum-materials-health-care-degree-programs-biosimilars; accessed on 4 April 2022) [9].
- Peptide mapping (LC-MS), peptide mass fingerprinting (MALDI-MS), MALDI TOF, and MS amino acid sequencing are all examples of primary structure sequencing.
- Higher-order structures can be confirmed using thermodynamic DSC, NMR, SPR, ELISA, fluorescence, far and near UV CD, DSC, NMR, SPR, and ELISA. While process-related testing is straightforward and well-established, testing product-related attributes can be improved by testing the UV and fluorescence spectra under various stress conditions, temperature surfactants, electrolytes, and pH [10]. Newer and more sensitive methods are always needed.
- Cell-based assays, SPR, and ELISA, to test receptor binding.
- Forced degradation: degradation is forced to match intramolecular bond strength as a structural similarity measure.
3. Process-Related Attributes
The process-related quality attributes are dependent on the manufacturing process used; thus, they are made part of the release specification to assure compliance. Establishing the acceptance criteria for these quality attributes can be achieved based on legacy values, as is considered to be standard practice for injectable products or the criteria established by testing the reference product.
Ideally, a process-related attribute should be made part of the release specification. The release limits can be derived from legacy values (previously established and known) or by testing the reference product. The European and British Pharmacopoeias [11] have developed monographs of several key biological products defining quality attributes to establish release specifications. The USP has stated that it will not develop monographs for a biologic unless there is stakeholder consensus supporting its creation, including the support of the FDA [12]. The FDA has discouraged the USP from creating biologics monographs to ensure that innovator biologics makers do not use the monograph process to block biosimilar competition by incorporating patented characteristics of their product that are not relevant to safety, purity, or potency, thereby further impacting competition [13].
However, despite the different opinions on using a monograph to develop a biosimilar product, many legacy attributes, the quality attributes that come from historical and experience-based variability, are widely accepted as norms.
- Protein content. Biological products label potency of 100 IU/mL for insulin in vials. Based on shared experience, the protein content cannot always be the same due to filling variability, concentration testing variability, and many other unpredictable factors. For this reason, most products are allowed an acceptable practical range of variability of ±5% [14]. However, this quality attribute is controversial, as the first FDA guideline required this attribute to be tested for equivalence. The 95% CI of the biosimilar product cannot go beyond 1.5*SD of the reference product in an equivalence test. This range was established entirely arbitrarily. If the SD of the reference product turns out to be small, all batches of the biosimilar product will fail despite being within the release specification of ±5%. This means that a biosimilar product might be acceptable for patients but not for approval by the FDA. This situation arose when the first biosimilar EP2006 required the testing of 50 lots to match the equivalence criteria of Amgen’s Neupogen, despite all lots meeting the release specifications [15]. We can use this as an example to remove the comparative testing of the protein content from side-by-side testing. However, if a biosimilar product has a higher variability, this must be confirmed with the variability in the reference product lots.
- Post-translation modifications, aggregates, and isomers should be tested in a range model, wherein 90% of the values of the biosimilar lots should fall within 3 × SD of the reference product to establish analytical similarity and the specification should include a range of no more than 3 × SD of the reference product.
- Bioassay limits are calculated as specified in the statistical analysis of biological assays and test results. They are typically expressed as an acceptable range for the estimated potency (e.g., 80–125 percent of the stated potency) and an acceptable range for the confidence limits of the estimated potency (e.g., 64–156 percent of the stated potency) [16].
- Impurities in biological products, also known as residuals, are of much greater importance than in chemical drugs. Impurities can be either process- or product-related. Process-related impurities are derived from the manufacturing process—for example, cell culture, downstream, or cell substrates. In contrast, product-related impurities are non-active molecular variants of the biologic and are formed during expression, manufacture, or storage. Understanding these impurities is essential to developing control strategies to reduce or remove them from the final product. The impurities caused by the upstream process may include cell culture reagents, antifoams, growth modifiers (insulin), antibiotics, protein a, solubilizers, residual solvents, chelating agents, extractable extracts, and leachable. The downstream-derived impurities may include detergent, protein a, process additives, chromatographic resins, extractable, and leachable. Cell-derived impurities include host cell DNA and host cell proteins. Product-related impurities include truncated forms such as fragments; modified forms such as disulfide, oxidation, deamidation, and glycosylation; and aggregates including multimers and subvisible particles. When present in a substantial quantity, these impurities may reduce the product’s potency and, worse, induce immunogenic responses or alter the product’s pharmacokinetics. While process-related impurities can be readily isolated, product-related impurities are often difficult to separate because of their close structural similarity to the active molecules. As a result, a biosimilar product must not have any unmatched impurity. There is also no analytical method or biological test that can ensure the safety of an unmatched impurity since any testing of immunogenicity in an animal species may not match the immune response in humans. In some cases, an unmatched impurity may be acceptable if the same regulatory agency has approved an identical structure or there is sufficient published proof of its safety. Since matched impurities can reduce efficacy if they are not as efficacious, a variation of 3% is generally allowed as a legacy attribute. Additionally, the 3% variation must not include more than 1% of any single impurity. However, these acceptance criteria can also be established by profiling the reference product.
- Particle size (subvisible), residual DNA, fill volume, and sterility standards are well defined in several official compendia, and these should be acceptable.
- Physical properties. If the formulation is the same, then the formulation’s physical properties, such as surfactants, osmolality, and pH, should fall within three standard deviations of that of the reference product. However, when the formulation is different, the release specifications will be based on testing multiple lots of biosimilar products. The BPCIA allows a biosimilar product to have a different formulation; however, using the same formulation as the reference product reduces the risk of higher immunogenicity, especially if the inactive component(s) are used in another biological product and have the same route of administration. This is in contrast to the WHO’s suggestion that “relevant differences in formulation (for example, use of excipients in the biosimilar that are not widely used in medicinal products)” can be tested using animal models [17], despite experience gained from the incidence of immunological reactions induced by erythropoietin formulations that used a different formulation [18]. No animal testing can establish the safety of inactive components when used in a biological drug formulation.
Since analytical similarity assessment is the core of biosimilar product evaluation, most regulatory audits pertain to these details after filing the registration application. They often result in multiple complete response letters (CRLs) that delay approval. This includes data integrity and CFR 21 Part 11 compliance, proof of test method suitability (suitable or validated), and blinding issues. Therefore, outsourcing the analytical assessment may be more cost-effective and time-effective. First, this realization regarding the analytical assessment audits came after multiple products were filed, and several qualified CDMOs can now fulfill this role.
References
- FDA. Guideline on Statistical Approaches to Evaluate Analytical Similarity Guidance for Industry. 2017. Available online: https://www.pbwt.com/content/uploads/2018/06/UCM576786.pdf (accessed on 23 March 2022).
- FDA Withdraws Draft Guidance for Industry: Statistical Approaches to Evaluate Analytical Similarity. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-withdraws-draft-guidance-industry-statistical-approaches-evaluate-analytical-similarity (accessed on 23 March 2022).
- FDA. Development of Therapeutic Protein Biosimilars: Comparative Analytical Assessment and Other Quality-Related Considerations Guidance for Industry. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/development-therapeutic-protein-biosimilars-comparative-analytical-assessment-and-other-quality (accessed on 23 March 2022).
- Forbes Magazine. One Man’s Mission to Fix the FDA’s Biosimilar Problem. Available online: https://www.forbes.com/sites/nicolefisher/2018/07/25/one-mans-mission-to-fix-the-fdas-biosimilar-problem/?sh=1843e1723808 (accessed on 23 March 2022).
- Wolff-Holz, E.; Tiitso, K.; Vleminckx, C.; Weise, M. Evolution of the EU Biosimilar Framework: Past and Future. BioDrugs 2019, 33, 621–634.
- Niazi, S. Analysis of FDA-Licensed Biosimilars: Time for a Paradigm Shift. AJMC, Center for Biosimilars. Available online: https://www.centerforbiosimilars.com/view/analysis-of-fda-licensed-biosimilars-time-for-a-paradigm-shift (accessed on 23 March 2022).
- European Medicines Agency Biotechnology Products. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-biotechnology-derived-proteins-active_en-0.pdf (accessed on 23 March 2022).
- Vandekerckhove, K.; Seidl, A.; Gutka, H.; Kumar, M.; Gratzl, G.; Keire, D.; Coffey, T.; Kuehne, H. Rational Selection, Criticality Assessment, and Tiering of Quality Attributes and Test Methods for Analytical Similarity Evaluation of Biosimilars. AAPS J. 2018, 20, 68.
- FDA. Level 1 Biosimilar and Interchangeable Products Foundational Concepts. Available online: https://www.fda.gov/drugs/biosimilars/curriculum-materials-health-care-degree-programs-biosimilars (accessed on 23 March 2022).
- Niazi, S. Methods for Comparing a Structure of a First Biomolecule and a Second Biomolecule. U.S. Patent 20,140,356,968, 4 December 2014. Available online: https://tinyurl.com/h58tdjnr (accessed on 23 March 2022).
- European Directorate for the Quality of Medicines & HealthCare. Biotherapeutics Monographs. Available online: https://www.edqm.eu/sites/default/files/medias/bio_tab_portfolio_january_2022.pdf (accessed on 4 April 2022).
- USP. Statement on Monographs for Biologics. US Pharmacopoeia. Available online: https://www.usp.org/news/statement-on-monographs-for-biologics (accessed on 23 March 2022).
- FDA-USP Clash over Biologics Monographs. Available online: https://www.raps.org/news-and-articles/news-articles/2019/6/fda-usp-clash-over-biologics-monographs (accessed on 4 April 2022).
- Goyal, P.; Pai, H.V.; Kodali, P.; Vats, B.; Vajpai, N.; Annegowda, S.; Mane, K.; Mohan, S.; Saxena, S.; Veerabhadraia, A.B.; et al. Physicochemical and functional characterization of MYL-1501D, a proposed biosimilar to insulin glargine. PLoS ONE 2021, 16, e0253168.
- FDA. Oncology Briefing Document. 15 January 2015. Available online: https://patentdocs.typepad.com/files/briefing-document.pdf (accessed on 23 March 2022).
- European Directorate for the Quality of Medicines & HealthCare. Synthetic Peptides and rDNA Products. Available online: https://www.edqm.eu/sites/default/files/guide_ph_eur_synthetic_peptides_and_rdna_proteins_2018.pdf (accessed on 23 March 2022).
- World Health Organization. WHO Guidelines on Evaluation of Biosimilars. Available online: https://cdn.who.int/media/docs/default-source/biologicals/who-guidelines-on-evaluation-of-biosimilars---4-nov-2021.pdf?sfvrsn=f17799ae_5 (accessed on 23 March 2022).
- McKoy, J.M.; Stonecash, R.E.; Cournoyer, D.; Rossert, J.; Nissenson, A.R.; Raisch, D.W.; Casadevall, N.; Bennett, C.L. Epoetin-associated pure red cell aplasia: Past, present, and future considerations. Transfusion 2008, 48, 1754–1762.
More
Information
Subjects:
Pharmacology & Pharmacy
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.4K
Entry Collection:
Biopharmaceuticals Technology
Revisions:
3 times
(View History)
Update Date:
27 Apr 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No