 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Agapios Sachinidis | -- | 2690 | 2022-04-26 16:00:28 | | | |
| 2 | Jessie Wu | Meta information modification | 2690 | 2022-04-27 04:40:45 | | |
Video Upload Options
Induced pluripotent stem cell (iPSC)-based disease modelling and the cell replacement therapy approach have proven to be very powerful and instrumental in biomedical research and personalized regenerative medicine as evidenced in the past decade by unraveling novel pathological mechanisms of a multitude of monogenic diseases at the cellular level and the ongoing and emerging clinical trials with iPSC-derived cell products. iPSC-based disease modelling has sparked widespread enthusiasm and has presented an unprecedented opportunity in high throughput drug discovery platforms and safety pharmacology in association with three-dimensional multicellular organoids such as personalized organs-on-chips, gene/base editing, artificial intelligence and high throughput “omics” methodologies.
1. Induced Pluripotent Stem Cell-Based Disease Modeling:
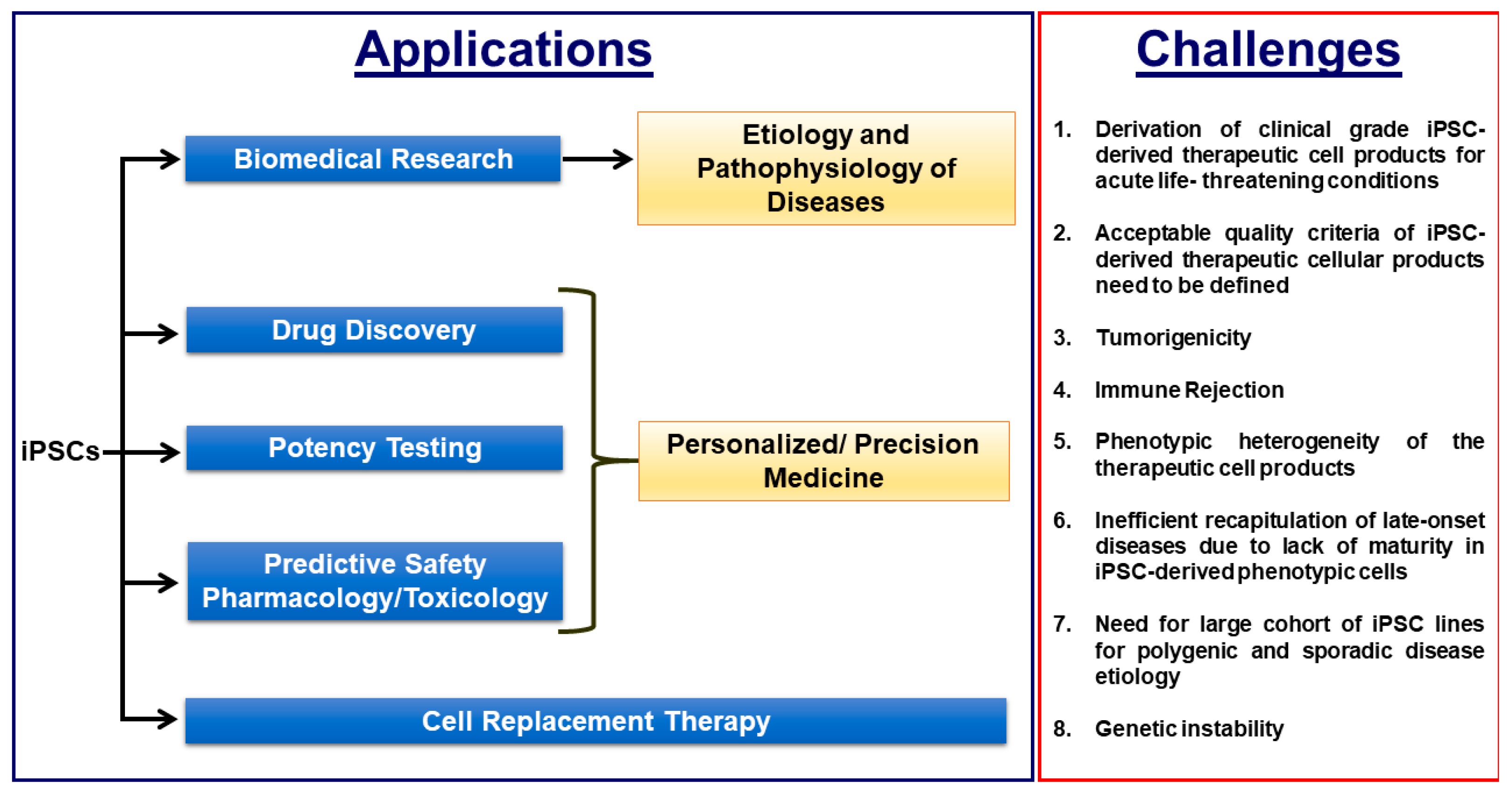
2. Derivation of Clinical Grade iPSCs and Biobanking of Universal Cell Lines
3. Defining the Quality Attributes of Good iPSCs and Their Differentiated Therapeutic Cellular Products
| S.No | Quality Attributes | iPSCs | iPSC-Derived Differentiated Therapeutic Product |
|---|---|---|---|
|
1 |
Sterility and free of mycoplasma and endotoxins as required by the cGMP guidelines |
✓ |
✓ |
|
2 |
Expression of pluripotency associated marks such as NANOG, OCT4, SSEA-3, SSEA-4, TRA-1-60, TRA-1-81, SOX2 [Pluritest™, hPSC Scorecard™] |
✓ |
✕ |
|
3 |
Expression of differentiation markers unique to the therapeutic cellular product |
✓ |
|
|
4 |
Normal Karyotype and Absence of chromosomal aberrations |
✓ |
✓ |
|
5 |
Absence of undifferentiated iPSC in the final cellular drug product and free of tumorigenicity as analysed by: A. in vivo teratoma assay B. Whole Genome and Exome Sequencing with cancer associated gene panels C. Flow cytometry with the panel of cancer associated markers |
✕ |
✓ |
|
6 |
100 % purity of the therapeutic cellular product without any contaminating other lineage cell types such as neuronal cells and hepatic cells and other cell subtypes such atrial and pacemaker cell types in therapeutic ventricular cell product, for example |
✕ |
✓ |
|
7 |
Supporting in vivo data on the cell engraftment, durabity and functional improvement in pre-clinical models |
✕ |
✓ |
|
8 |
Absence of residual reprogramming transgenes and vectors by Whole Genome and Exome Sequencing |
✓ |
✓ |
|
9 |
Genotyping in case of autologous iPSCs approach [ Short Tandem Repeat Analysis] |
✓ |
✓ |
|
10 |
Viability |
✓ |
✓ |
4. Tumorigenicity
5. Immune Rejection
6. The Conundrum of Choosing Allogenic or Autologous iPSCs for More Efficient Cell Therapy
References
- Gunaseeli, I.; Doss, M.X.; Antzelevitch, C.; Hescheler, J.; Sachinidis, A. Induced pluripotent stem cells as a model for accelerated patient- and disease-specific drug discovery. Curr. Med. Chem. 2010, 17, 759–766.
- van den Berg, A.; Mummery, C.L.; Passier, R.; van der Meer, A.D. Personalised organs-on-chips: Functional testing for precision medicine. Lab. Chip 2019, 19, 198–205.
- Schork, N.J.; Nazor, K. Integrated Genomic Medicine: A Paradigm for Rare Diseases and Beyond. Adv. Genet. 2017, 97, 81–113.
- Bouvy, J.C.; De Bruin, M.L.; Koopmanschap, M.A. Epidemiology of adverse drug reactions in Europe: A review of recent observational studies. Drug Saf. 2015, 38, 437–453.
- Gaspar, J.A.; Doss, M.X.; Hengstler, J.G.; Cadenas, C.; Hescheler, J.; Sachinidis, A. Unique metabolic features of stem cells, cardiomyocytes, and their progenitors. Circ. Res. 2014, 114, 1346–1360.
- Stricker, S.; Pollard, S. Reprogramming cancer cells to pluripotency: An experimental tool for exploring cancer epigenetics. Epigenetics 2014, 9, 798–802.
- Stricker, S.H.; Feber, A.; Engstrom, P.G.; Caren, H.; Kurian, K.M.; Takashima, Y.; Watts, C.; Way, M.; Dirks, P.; Bertone, P.; et al. Widespread resetting of DNA methylation in glioblastoma-initiating cells suppresses malignant cellular behavior in a lineage-dependent manner. Gene Dev. 2013, 27, 654–669.
- Miyoshi, K.; Tsuji, D.; Kudoh, K.; Satomura, K.; Muto, T.; Itoh, K.; Noma, T. Generation of human induced pluripotent stem cells from oral mucosa. J. Biosci. Bioeng. 2010, 110, 345–350.
- Miyazaki, S.; Yamamoto, H.; Miyoshi, N.; Wu, X.; Ogawa, H.; Uemura, M.; Nishimura, J.; Hata, T.; Takemasa, I.; Mizushima, T.; et al. A Cancer Reprogramming Method Using MicroRNAs as a Novel Therapeutic Approach against Colon Cancer: Research for Reprogramming of Cancer Cells by MicroRNAs. Ann. Surg. Oncol. 2015, 22, S1394–S1401.
- Crespo, M.; Vilar, E.; Tsai, S.Y.; Chang, K.; Amin, S.; Srinivasan, T.; Zhang, T.; Pipalia, N.H.; Chen, H.J.; Witherspoon, M.; et al. Colonic organoids derived from human induced pluripotent stem cells for modeling colorectal cancer and drug testing. Nat. Med. 2017, 23, 878–884.
- Gingold, J.; Zhou, R.; Lemischka, I.R.; Lee, D.F. Modeling Cancer with Pluripotent Stem Cells. Trends Cancer 2016, 2, 485–494.
- Kim, H.; Yoo, S.; Zhou, R.; Xu, A.; Bernitz, J.M.; Yuan, Y.; Gomes, A.M.; Daniel, M.G.; Su, J.; Demicco, E.G.; et al. Oncogenic role of SFRP2 in p53-mutant osteosarcoma development via autocrine and paracrine mechanism. Proc. Natl. Acad. Sci. USA 2018, 115, E11128–E11137.
- Kotini, A.G.; Chang, C.J.; Boussaad, I.; Delrow, J.J.; Dolezal, E.K.; Nagulapally, A.B.; Perna, F.; Fishbein, G.A.; Klimek, V.M.; Hawkins, R.D.; et al. Functional analysis of a chromosomal deletion associated with myelodysplastic syndromes using isogenic human induced pluripotent stem cells. Nat. Biotechnol. 2015, 33, 646–655.
- Kotini, A.G.; Chang, C.J.; Chow, A.; Yuan, H.; Ho, T.C.; Wang, T.; Vora, S.; Solovyov, A.; Husser, C.; Olszewska, M.; et al. Stage-Specific Human Induced Pluripotent Stem Cells Map the Progression of Myeloid Transformation to Transplantable Leukemia. Cell Stem Cell 2017, 20, 315–328.e317.
- Lee, D.F.; Su, J.; Kim, H.S.; Chang, B.; Papatsenko, D.; Zhao, R.; Yuan, Y.; Gingold, J.; Xia, W.; Darr, H.; et al. Modeling familial cancer with induced pluripotent stem cells. Cell 2015, 161, 240–254.
- Mulero-Navarro, S.; Sevilla, A.; Roman, A.C.; Lee, D.F.; D’Souza, S.L.; Pardo, S.; Riess, I.; Su, J.; Cohen, N.; Schaniel, C.; et al. Myeloid Dysregulation in a Human Induced Pluripotent Stem Cell Model of PTPN11-Associated Juvenile Myelomonocytic Leukemia. Cell Rep. 2015, 13, 504–515.
- Papapetrou, E.P. Patient-derived induced pluripotent stem cells in cancer research and precision oncology. Nat. Med. 2016, 22, 1392–1401.
- Sommer, C.A.; Capilla, A.; Molina-Estevez, F.J.; Gianotti-Sommer, A.; Skvir, N.; Caballero, I.; Chowdhury, S.; Mostoslavsky, G. Modeling APC mutagenesis and familial adenomatous polyposis using human iPS cells. PLoS ONE 2018, 13, e0200657.
- Zhou, R.; Xu, A.; Gingold, J.; Strong, L.C.; Zhao, R.; Lee, D.F. Li-Fraumeni Syndrome Disease Model: A Platform to Develop Precision Cancer Therapy Targeting Oncogenic p53. Trends Pharmacol. Sci. 2017, 38, 908–927.
- Curry, E.L.; Moad, M.; Robson, C.N.; Heer, R. Using induced pluripotent stem cells as a tool for modelling carcinogenesis. World J. Stem Cells 2015, 7, 461–469.
- Esch, E.W.; Bahinski, A.; Huh, D. Organs-on-chips at the frontiers of drug discovery. Nat. Rev. Drug Discov. 2015, 14, 248–260.
- Low, L.A.; Tagle, D.A. Organs-on-chips: Progress, challenges, and future directions. Exp. Biol. Med. (Maywood) 2017, 242, 1573–1578.
- Liu, C.; Oikonomopoulos, A.; Sayed, N.; Wu, J.C. Modeling human diseases with induced pluripotent stem cells: From 2D to 3D and beyond. Development 2018, 145.
- Ribas, J.; Zhang, Y.S.; Pitrez, P.R.; Leijten, J.; Miscuglio, M.; Rouwkema, J.; Dokmeci, M.R.; Nissan, X.; Ferreira, L.; Khademhosseini, A. Biomechanical Strain Exacerbates Inflammation on a Progeria-on-a-Chip Model. Small 2017, 13.
- Atchison, L.; Zhang, H.; Cao, K.; Truskey, G.A. A Tissue Engineered Blood Vessel Model of Hutchinson-Gilford Progeria Syndrome Using Human iPSC-derived Smooth Muscle Cells. Sci. Rep. 2017, 7, 8168.
- Aasen, T.; Raya, A.; Barrero, M.J.; Garreta, E.; Consiglio, A.; Gonzalez, F.; Vassena, R.; Bilic, J.; Pekarik, V.; Tiscornia, G.; et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat. Biotechnol. 2008, 26, 1276–1284.
- Loh, Y.H.; Agarwal, S.; Park, I.H.; Urbach, A.; Huo, H.; Heffner, G.C.; Kim, K.; Miller, J.D.; Ng, K.; Daley, G.Q. Generation of induced pluripotent stem cells from human blood. Blood 2009, 113, 5476–5479.
- Dambrot, C.; van de Pas, S.; van Zijl, L.; Brandl, B.; Wang, J.W.; Schalij, M.J.; Hoeben, R.C.; Atsma, D.E.; Mikkers, H.M.; Mummery, C.L.; et al. Polycistronic lentivirus induced pluripotent stem cells from skin biopsies after long term storage, blood outgrowth endothelial cells and cells from milk teeth. Differentiation 2013, 85, 101–109.
- Seki, T.; Yuasa, S.; Oda, M.; Egashira, T.; Yae, K.; Kusumoto, D.; Nakata, H.; Tohyama, S.; Hashimoto, H.; Kodaira, M.; et al. Generation of induced pluripotent stem cells from human terminally differentiated circulating T cells. Cell Stem Cell 2010, 7, 11–14.
- Seki, T.; Yuasa, S.; Fukuda, K. Derivation of induced pluripotent stem cells from human peripheral circulating T cells. Curr. Protoc. Stem Cell Biol. 2011, 4, Unit4A 3.
- Maherali, N.; Ahfeldt, T.; Rigamonti, A.; Utikal, J.; Cowan, C.; Hochedlinger, K. A high-efficiency system for the generation and study of human induced pluripotent stem cells. Cell Stem Cell 2008, 3, 340–345.
- Cao, Y.; Xu, J.; Wen, J.; Ma, X.; Liu, F.; Li, Y.; Chen, W.; Sun, L.; Wu, Y.; Li, S.; et al. Generation of a Urine-Derived Ips Cell Line from a Patient with a Ventricular Septal Defect and Heart Failure and the Robust Differentiation of These Cells to Cardiomyocytes via Small Molecules. Cell Physiol. Biochem. 2018, 50, 538–551.
- Gaignerie, A.; Lefort, N.; Rousselle, M.; Forest-Choquet, V.; Flippe, L.; Francois-Campion, V.; Girardeau, A.; Caillaud, A.; Chariau, C.; Francheteau, Q.; et al. Urine-derived cells provide a readily accessible cell type for feeder-free mRNA reprogramming. Sci. Rep. 2018, 8, 14363.
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290.
- Polo, J.M.; Liu, S.; Figueroa, M.E.; Kulalert, W.; Eminli, S.; Tan, K.Y.; Apostolou, E.; Stadtfeld, M.; Li, Y.; Shioda, T.; et al. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat. Biotechnol. 2010, 28, 848–855.
- Kim, K.; Zhao, R.; Doi, A.; Ng, K.; Unternaehrer, J.; Cahan, P.; Huo, H.; Loh, Y.H.; Aryee, M.J.; Lensch, M.W.; et al. Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 1117–1119.
- Bar-Nur, O.; Russ, H.A.; Efrat, S.; Benvenisty, N. Epigenetic memory and preferential lineage-specific differentiation in induced pluripotent stem cells derived from human pancreatic islet beta cells. Cell Stem Cell 2011, 9, 17–23.
- Boland, M.J.; Nazor, K.L.; Loring, J.F. Epigenetic regulation of pluripotency and differentiation. Circ. Res. 2014, 115, 311–324.
- Ghosh, Z.; Wilson, K.D.; Wu, Y.; Hu, S.; Quertermous, T.; Wu, J.C. Persistent donor cell gene expression among human induced pluripotent stem cells contributes to differences with human embryonic stem cells. PLoS ONE 2010, 5, e8975.
- Nishino, K.; Toyoda, M.; Yamazaki-Inoue, M.; Fukawatase, Y.; Chikazawa, E.; Sakaguchi, H.; Akutsu, H.; Umezawa, A. DNA methylation dynamics in human induced pluripotent stem cells over time. PLoS Genet. 2011, 7, e1002085.
- Lo Sardo, V.; Ferguson, W.; Erikson, G.A.; Topol, E.J.; Baldwin, K.K.; Torkamani, A. Influence of donor age on induced pluripotent stem cells. Nat. Biotechnol. 2017, 35, 69–74.
- Musunuru, K.; Sheikh, F.; Gupta, R.M.; Houser, S.R.; Maher, K.O.; Milan, D.J.; Terzic, A.; Wu, J.C.; American Heart Association Council on Functional, G.; Translational, B.; et al. Induced Pluripotent Stem Cells for Cardiovascular Disease Modeling and Precision Medicine: A Scientific Statement From the American Heart Association. Circ. Genom. Precis. Med. 2018, 11, e000043.
- Ban, H.; Nishishita, N.; Fusaki, N.; Tabata, T.; Saeki, K.; Shikamura, M.; Takada, N.; Inoue, M.; Hasegawa, M.; Kawamata, S.; et al. Efficient generation of transgene-free human induced pluripotent stem cells (iPSCs) by temperature-sensitive Sendai virus vectors. Proc. Natl. Acad. Sci. USA 2011, 108, 14234–14239.
- Narsinh, K.H.; Jia, F.; Robbins, R.C.; Kay, M.A.; Longaker, M.T.; Wu, J.C. Generation of adult human induced pluripotent stem cells using nonviral minicircle DNA vectors. Nat. Protoc. 2011, 6, 78–88.
- Stadtfeld, M.; Nagaya, M.; Utikal, J.; Weir, G.; Hochedlinger, K. Induced pluripotent stem cells generated without viral integration. Science 2008, 322, 945–949.
- Warren, L.; Ni, Y.; Wang, J.; Guo, X. Feeder-free derivation of human induced pluripotent stem cells with messenger RNA. Sci. Rep. 2012, 2, 657.
- Pfaff, N.; Fiedler, J.; Holzmann, A.; Schambach, A.; Moritz, T.; Cantz, T.; Thum, T. miRNA screening reveals a new miRNA family stimulating iPS cell generation via regulation of Meox2. EMBO Rep. 2011, 12, 1153–1159.
- Yu, J.; Hu, K.; Smuga-Otto, K.; Tian, S.; Stewart, R.; Slukvin, I.I.; Thomson, J.A. Human induced pluripotent stem cells free of vector and transgene sequences. Science 2009, 324, 797–801.
- Okita, K.; Matsumura, Y.; Sato, Y.; Okada, A.; Morizane, A.; Okamoto, S.; Hong, H.; Nakagawa, M.; Tanabe, K.; Tezuka, K.; et al. A more efficient method to generate integration-free human iPS cells. Nat. Methods 2011, 8, 409–412.
- Woltjen, K.; Michael, I.P.; Mohseni, P.; Desai, R.; Mileikovsky, M.; Hamalainen, R.; Cowling, R.; Wang, W.; Liu, P.; Gertsenstein, M.; et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature 2009, 458, 766–770.
- Attwood, S.W.; Edel, M.J. iPS-Cell Technology and the Problem of Genetic Instability-Can It Ever Be Safe for Clinical Use? J. Clin. Med. 2019, 8, 288.
- Miki, K.; Endo, K.; Takahashi, S.; Funakoshi, S.; Takei, I.; Katayama, S.; Toyoda, T.; Kotaka, M.; Takaki, T.; Umeda, M.; et al. Efficient Detection and Purification of Cell Populations Using Synthetic MicroRNA Switches. Cell Stem Cell 2015, 16, 699–711.
- Ito, E.; Miyagawa, S.; Takeda, M.; Kawamura, A.; Harada, A.; Iseoka, H.; Yajima, S.; Sougawa, N.; Mochizuki-Oda, N.; Yasuda, S.; et al. Tumorigenicity assay essential for facilitating safety studies of hiPSC-derived cardiomyocytes for clinical application. Sci. Rep. 2019, 9, 1881.
- Sullivan, S.; Stacey, G.N.; Akazawa, C.; Aoyama, N.; Baptista, R.; Bedford, P.; Bennaceur Griscelli, A.; Chandra, A.; Elwood, N.; Girard, M.; et al. Quality control guidelines for clinical-grade human induced pluripotent stem cell lines. Regen. Med. 2018, 13, 859–866.
- Knoepfler, P.S. Key anticipated regulatory issues for clinical use of human induced pluripotent stem cells. Regen. Med. 2012, 7, 713–720.
- Okita, K.; Ichisaka, T.; Yamanaka, S. Generation of germline-competent induced pluripotent stem cells. Nature 2007, 448, 313–317.
- Eschenhagen, T.; Weinberger, F. Heart Repair With Myocytes. Circ. Res. 2019, 124, 843–845.
- Garreta, E.; Sanchez, S.; Lajara, J.; Montserrat, N.; Belmonte, J.C.I. Roadblocks in the Path of iPSC to the Clinic. Curr. Transplant. Rep. 2018, 5, 14–18.
- de Almeida, P.E.; Meyer, E.H.; Kooreman, N.G.; Diecke, S.; Dey, D.; Sanchez-Freire, V.; Hu, S.; Ebert, A.; Odegaard, J.; Mordwinkin, N.M.; et al. Transplanted terminally differentiated induced pluripotent stem cells are accepted by immune mechanisms similar to self-tolerance. Nat. Commun. 2014, 5, 3903.
- Zhao, T.; Zhang, Z.N.; Westenskow, P.D.; Todorova, D.; Hu, Z.; Lin, T.; Rong, Z.; Kim, J.; He, J.; Wang, M.; et al. Humanized Mice Reveal Differential Immunogenicity of Cells Derived from Autologous Induced Pluripotent Stem Cells. Cell Stem Cell 2015, 17, 353–359.
- Huang, K.; Liu, P.; Li, X.; Chen, S.; Wang, L.; Qin, L.; Su, Z.; Huang, W.; Liu, J.; Jia, B.; et al. Neural progenitor cells from human induced pluripotent stem cells generated less autogenous immune response. Sci. China Life Sci. 2014, 57, 162–170.
- Mandai, M.; Watanabe, A.; Kurimoto, Y.; Hirami, Y.; Morinaga, C.; Daimon, T.; Fujihara, M.; Akimaru, H.; Sakai, N.; Shibata, Y.; et al. Autologous Induced Stem-Cell-Derived Retinal Cells for Macular Degeneration. N. Engl. J. Med. 2017, 376, 1038–1046.
- Gornalusse, G.G.; Hirata, R.K.; Funk, S.E.; Riolobos, L.; Lopes, V.S.; Manske, G.; Prunkard, D.; Colunga, A.G.; Hanafi, L.A.; Clegg, D.O.; et al. HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat. Biotechnol. 2017, 35, 765–772.
- Mattapally, S.; Pawlik, K.M.; Fast, V.G.; Zumaquero, E.; Lund, F.E.; Randall, T.D.; Townes, T.M.; Zhang, J. Human Leukocyte Antigen Class I and II Knockout Human Induced Pluripotent Stem Cell-Derived Cells: Universal Donor for Cell Therapy. J. Am. Heart Assoc. 2018, 7, e010239.
- Deuse, T.; Hu, X.; Gravina, A.; Wang, D.; Tediashvili, G.; De, C.; Thayer, W.O.; Wahl, A.; Garcia, J.V.; Reichenspurner, H.; et al. Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat. Biotechnol. 2019, 37, 252–258.
- Ohnuki, M.; Takahashi, K. Present and future challenges of induced pluripotent stem cells. Philos Trans. R Soc. Lond. B Biol. Sci. 2015, 370, 20140367.
- Bravery, C.A. Do human leukocyte antigen-typed cellular therapeutics based on induced pluripotent stem cells make commercial sense? Stem Cells Dev. 2015, 24, 1–10.
- Jacquet, L.; Stephenson, E.; Collins, R.; Patel, H.; Trussler, J.; Al-Bedaery, R.; Renwick, P.; Ogilvie, C.; Vaughan, R.; Ilic, D. Strategy for the creation of clinical grade hESC line banks that HLA-match a target population. EMBO Mol. Med. 2013, 5, 10–17.


