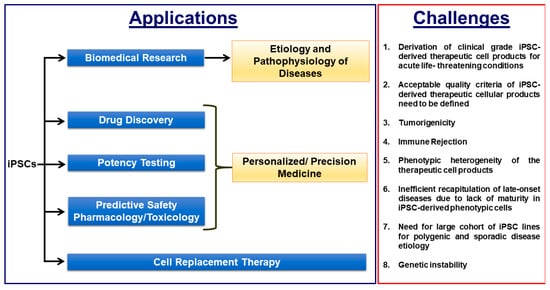
Induced pluripotent stem cell (iPSC)-based disease modelling and the cell replacement therapy approach have proven to be very powerful and instrumental in biomedical research and personalized regenerative medicine as evidenced in the past decade by unraveling novel pathological mechanisms of a multitude of monogenic diseases at the cellular level and the ongoing and emerging clinical trials with iPSC-derived cell products. iPSC-based disease modelling has sparked widespread enthusiasm and has presented an unprecedented opportunity in high throughput drug discovery platforms and safety pharmacology in association with three-dimensional multicellular organoids such as personalized organs-on-chips, gene/base editing, artificial intelligence and high throughput “omics” methodologies.
1. Induced Pluripotent Stem Cell-Based Disease Modeling:
The major critical component in delineating the etiology and pathophysiology of any human disease and drug discovery is the requirement of a physiologically relevant experimental model of disease, either
in vitro or
in vivo or both, that faithfully recapitulates the respective pathophysiology and clinical manifestations. To this end, animal modelling, most frequently with mice, has been the key player in both basic research and pharmaceutical research and development (R&D) as a non-clinical efficacy model [
18]. More often, the translation of drug trials to humans from experimental animal models fails due to species differences in biological responses, leading to high failure rates as reflected by the number of new drugs approved by the US Food and Drug Administration for clinical use every year. Therefore, it is more appealing to use an appropriate human model of diseases
in vitro that recapitulates the pathophysiological mechanisms. Human primary cell-based disease modelling would be helpful but will be limited due to insufficient expandable cellular sources from patients, in particular hard to access cells such as cardiomyocytes, neuronal cells, pancreatic beta cells and other clinically relevant cells from organs other than skin and peripheral blood.
The so called “personalized medicine” approach in which each individual patient would receive a tailored treatment is becoming critically important in medicine, pharmacology and toxicology to overcome possible adverse side effects and to minimize the frequency of non-responders. For example, the total number of people required to take a drug in order for only one of them to benefit from its effects ranges from 5–50 for some of the highest-grossing drugs, implying the high frequency of non-responders for certain drugs [
20,
21]. Even more alarmingly, millions of people are hospitalized annually due to adverse side effects due to their medications, resulting in hundreds of thousands of deaths per year and highlighting the critical need for precision medicine to eliminate the adverse mortality and morbidity due to side effects of drugs [
20,
22]. To this end, human induced pluripotent stem cell (iPSC)-based disease models (
Figure 2) are a promising candidate due to their unlimited supply of clinically relevant phenotypic cells, their human origin, their derivation potential from any individual, easy accessibility and scalability and the considerable advances in understanding the etiology and progression of a diverse array of diseases such as Parkinson’s disease, Alzheimer’s disease and inherited cardiac diseases. A side-by-side comparison of human iPSC based
in vitro and rodent
in vivo experimental models will synergistically accelerate elucidating the pathophysiology mechanisms of diseases in basic research, novel drug discovery and safety pharmacology in a unprecedentent manner.
Figure 1. Biomedical applications of iPSCs and the critical challenges that need to be overcome for efficient clinical translation.
Interestingly, iPSCs share a number of characteristics with cancer cells including indefinite proliferation capacity and the expression of oncogenic markers like c-MYC and metabolic signatures [
23]. The generation of iPSCs from human cancer cells represents an opportunity to develop
in vitro models of carcinogenesis for specific cancer types such as glioblastoma and gastrointestinal cancer, since the lack of a relevant model to study cancer progression has limited research which is suitable for translation to clinical settings [
24,
25,
26,
27]. Interestingly, iPSC disease models of several cancer-prone diseases such as Li–Fraumeni syndrome, Noonan syndrome, myelodysplastic syndromes, and familial adenomatous polyposis appear to be more attractive candidates in the study of cancer initiation and progression [
28,
29,
30,
31,
32,
33,
34,
35,
36,
37]. Also, iPSCs from somatic cells could be used to study carcinogenesis via overexpression or silencing of oncogenes and tumor suppressor genes and tracking the cellular changes and behaviors during cancer initiation and progression [
38].
Organs-on-chips are microfluidic cell culture systems seeded with patient specific iPSC-derived phenotypic cells that serve as functional units of human organs with a controlled, dynamic condition that recapitulates the complex tissue architecture and the physio-chemical microenvironment of tissues in the human body. These systems exhibit tissue- and organ-level functions that are not recapitulated in other 2D/3D
in vitro cell models [
20]. This organ-on-chip technology is being employed to develop more physiologically relevant, cost-effective
in vitro models for hit-to-lead and lead optimization that can more reliably predict the efficacy, toxicity and pharmacokinetics of drugs in humans [
39]. These chips are increasingly used as physiologically relevant pre-clinical disease models for a wide range of different diseases such as Barth syndrome-associated cardiomyopathy, dilated cardiomyopathy, drug-induced kidney glomerular injury, blood–brain barrier function and wound healing in drug discovery and safety pharmacology platforms [
20,
39,
40,
41]. Interestingly, the patient specific organs-on-chips made up of iPSCs derived from Hutchinson–Gilford progeria syndrome patients revealed exacerbated inflammation as well as reduced vasoactive function [
42,
43].
2. Derivation of Clinical Grade iPSCs and Biobanking of Universal Cell Lines
The first step in generating iPSCs is the selection of the donor cell type for the reprogramming process. iPSCs have been commonly generated from dermal fibroblasts from punch-skin biopsies, T cells from peripheral blood, renal tubular cells collected from urine samples and keratinocytes isolated from plucked hair [
15,
16,
56,
57,
58,
59,
60,
61]. Several studies have reported that iPSCs retain some degree of residual epigenetic memory from the somatic cell source from which they are derived and this can lead to their biased differential potential into certain cell types depending on the donor cell source due to the incomplete resetting of the non-CpG methylation patterns during reprogramming [
62,
63,
64,
65,
66]. However, it has been shown that their residual epigenetic memory diminishes as the cells are passaged in culture over a period of time [
67,
68]. An important concern on the choice of donor cell type is that some types of donor cells such as skin biopsy-derived dermal fibroblasts and blood cells might carry more mutational burdens and chromosomal abnormalities due to exposure to ultraviolet radiation and high cell turnover rates, especially from older donors [
69,
70]. The second step in the iPSC generation process is the selection of the optimal method for cellular delivery of the reprogramming factors and the optimal combination of reprogramming factors for the iPSC derivation. The earlier methods of reprogramming made use of the retroviral and lentiviral delivery methods to deliver the reprogramming factors. This raised concerns that these delivery methods would cause insertional inactivation of tumor suppressor genes and/or insertional activation of oncogenes and that the constitutive expression of the reprogramming factors would alter the iPSC characteristics and their differentiation potentials. This necessitated the use of transient, integration-free methods of delivering the reprogramming factors such as viral delivery/transient transfection methods with the use of either Sendai virus, adenovirus, episomal plasmids, minicircle plasmids, mini-intronic plasmids, PiggyBac transposons, synthetic modified mRNAs, or miRNAs [
71,
72,
73,
74,
75,
76,
77,
78]. Among these, Sendai virus, episomal DNAs and synthetic mRNAs are the commonly used approaches in basic and clinical research to derive integration-free iPSCs due to their relatively high efficiency and relative simplicity. The episomal plasmids and Sendai virus methods have been the preferred methods of choice for deriving clinical grade iPSCs (
Figure 1) [
70].
3. Defining the Quality Attributes of Good iPSCs and Their Differentiated Therapeutic Cellular Products
Given the huge variability across iPSC lines and their differentiated derivatives in terms of their differentiation potential, epigenetic status, tumorigenic potential, immunogenic potential, maturation characteristics, batch variability and co-occurrence of heterogenous populations of lineage subtypes and/or non-relevant cells as contaminating cell populations, the successful clinical outcome of the cell replacement therapy, in terms of efficacy and safety with these cells, largely and very critically rely on the acceptable quality criteria (
Table 1) for these cells prior to the transplantation procedure in the patients. Failure to detect oncogenic mutations in these iPSCs and their derived cellular products is not a guarantor of the non-tumorigenicity aspect of the iPSC-based therapies. Even if known a priori, it may be possible that oncogenic aberrations could evade detection by the current high throughput sequencing methodologies due to sequencing errors and other technical limitations [
79]. An important consideration is that the acceptable quality attributes of iPSCs and their differentiated derivatives used for clinical applications should be well defined for the safety and efficacy and this aspect is currently very poorly defined. Directed differentiation protocols and phenotypic selection based on cell surface antigens by magnetic- or flow cytometry-based sorting to yield a pure population of clinically relevant cells and strategies to improve the maturation characteristics and to gauge the magnitude of the maturation status of the cells are critically needed to improve the quality attributes of these cells for more effective cell-based regenerative therapy. A recent novel methodology making use of synthetic microRNA switches to purify the cell populations will help to improve the purity of the cell populations even if the cell surface markers are not available to tag the relevant cells [
80]. The development of a consistent and reliable translational model and iPSC-derived clinical cellular products with well-defined cellular characteristics is therefore a vital pre-requisite for high throughput therapeutic applications and cell replacement therapy.
Table 1 lists the minimal quality attributes required for clinical grade iPSCs and their differentiated drug products [
81,
82].
Table 1. Minimal quality criteria required for clinical-grade iPSCs and their differentiated products.
| S.No |
Quality Attributes |
iPSCs |
iPSC-Derived Differentiated Therapeutic Product |
|
1
|
Sterility and free of mycoplasma and endotoxins as required by the cGMP guidelines
|
✓
|
✓
|
|
2
|
Expression of pluripotency associated marks such as NANOG, OCT4, SSEA-3, SSEA-4, TRA-1-60, TRA-1-81, SOX2 [Pluritest™, hPSC Scorecard™]
|
✓
|
✕
|
|
3
|
Expression of differentiation markers unique to the therapeutic cellular product
|
|
✓
|
|
4
|
Normal Karyotype and Absence of chromosomal aberrations
|
✓
|
✓
|
|
5
|
Absence of undifferentiated iPSC in the final cellular drug product and free of tumorigenicity as analysed by: A. in vivo teratoma assay
B. Whole Genome and Exome Sequencing with cancer associated gene panels C. Flow cytometry with the panel of cancer associated markers
|
✕
|
✓
|
|
6
|
100 % purity of the therapeutic cellular product without any contaminating other lineage cell types such as neuronal cells and hepatic cells and other cell subtypes such atrial and pacemaker cell types in therapeutic ventricular cell product, for example
|
✕
|
✓
|
|
7
|
Supporting in vivo data on the cell engraftment, durabity and functional improvement in pre-clinical models
|
✕
|
✓
|
|
8
|
Absence of residual reprogramming transgenes and vectors by Whole Genome and Exome Sequencing
|
✓
|
✓
|
|
9
|
Genotyping in case of autologous iPSCs approach [ Short Tandem Repeat Analysis]
|
✓
|
✓
|
|
10
|
Viability
|
✓
|
✓
|
4. Tumorigenicity
One of the major stumbling blocks in iPSC-based cell replacement therapy is the risk of potential tumorigenicity from undifferentiated iPSCs in the cell population that will be used for cellular transplantation. The key concept is that iPSCs will almost certainly never be used in regenerative medicine if they are not able to cause a teratoma in mice [
18,
83,
84]. iPSCs can form both teratomas and malignant tumors such as neuroblastoma and follicular carcinoma if transplanted in their undifferentiated pluripotent state
in vivo [
84]. Thus, the potential tumorigenicity risk to human patients from both teratomas and malignant tumors is quite possible if transplanted cells are contaminated with undifferentiated iPSCs. Although improved directed differentiation protocols, purification methods such as flow cytometry-/magnetic bead-based sorting and small chemical molecules that selectively cause cell death of undifferentiated iPSCs can eliminate the potential risk of tumorigenicity from potentially harmful undifferentiated iPSCs, it remains currently unknown whether these methods and tumorigenicity assays prior to transplantation are efficient and adequate enough to eliminate the risk of teratomas and malignant tumors upon transplantation in human subjects. Longer follow-up periods and very sensitive assays with the latest “-omics” approaches tailored to each and every individual patient’s genetic makeup are critically required on this end, along with analysis of cell survival, integration, durability, immune tolerance, cellular behavior, altered metabolism, functional improvements, undesired effects such as the risk of arrhythmogenicity in the case of myocardial infarction models and genetic stability in the target organs or respective tissues in the respective pre-clinical transplantation models [
85].
5. Immune Rejection
Earlier studies in mice reported that undifferentiated iPSCs were rejected in syngeneic recipients whereas recent studies report that the differentiated cells from iPSCs do not elicit immune rejection in syngeneic settings, implying that the differentiated cells may be less immunogenic compared to undifferentiated iPSCs [
86,
87,
88,
89]. However, this could differ between cell types. For example, in humanized mice, the iPSC-derived smooth muscle cells mounted immune rejection while retinal epithelial cells did not [
86,
87,
88,
89]. Of note are the recently published results on the first clinical trial with autologous iPSC-derived retinal epithelial cells in a patient with age-related macular degeneration who showed long-term survival of transplanted cells for 25 months without immune suppression [
19]. In the event that the allogenic cell therapy requires immunosuppression, there is a growing concern regarding the use of persistent immunosuppression that increases the risk of kidney failure, severe infections and tumors. The risk versus benefit ratio is highly debatable in this case. A universal immune tolerant iPSC line will be ideal to this end. Strategies to enable the allogenic iPSCs to evade both T cell- and natural killer cell-mediated immune responses have been reported [
90,
91]. A recently reported novel approach in which inactivation of major histocompatibility complex (MHC) class I and II genes and overexpression of CD47 in iPSCs enabled them to evade immune rejection in fully MHC mismatched allogeneic recipients and the transplanted cells survived long term without the use of immunosuppression [
92]. These immune escape approaches have the potential to open the door to allogeneic iPSC-derived cell products without immune rejection concerns and complications.
6. The Conundrum of Choosing Allogenic or Autologous iPSCs for More Efficient Cell Therapy
Despite the potential benefits of autologous iPSC therapies, there are also some limitations and challenges that need to be overcome. First, production of the clinical grade autologous iPSC -derived phenotypic cells requires a high production cost associated with individual patient-specific iPSC derivation by reprogramming along with clinical grade phenotypic cells derivation by differentiation with a stringent quality-controlled system. Second, the production of clinical grade human iPSC-derived clinically relevant phenotypic cells in a quality controlled and robust system, for example cardiomyocytes, can take approximately four months from start to finish. This can be critical since cardiac cell transplantation in chronic myocardial infarction will be less efficient than in subacute conditions [
85]. More often, it may not be possible to meet the deadline for effective treatments of some disease conditions such as spinal cord injuries [
6]. Therefore, the practical approach to take forward with iPSC-based cell therapy as most expert reviews conclude is the use of allogeneic iPSC-derived cell sources as opposed to autologous iPSCs with the major arguments that: (1) biobanking of a limited number of approved iPSC lines from various human leukocyte antigen (HLA)-homozygous donors that would match the majority of the population will be more efficient to provide large quantities of transplantable cells as an off-the-shelf-product from a quality controlled and rigorously tested production process; (2) regulatory clearance would be easier since any single line of such an iPSC bank could be thoroughly tested to be free of viral contamination, tumorigenicity and genome instability; and (3) the patient could more effectively benefit from the off-the-shelf product more readily in critical subacute conditions such as myocardial infarction and spinal cord injury. According to a recent estimate, it is likely to cost approximately $800,000 to produce a clinical grade autologous iPSC-derived cellular product in compliance with current good manufacturing practice (cGMP) requirements alone and approximately $10,000 to $20,000 for iPSC line generation for transplantation [
93,
94]. The allogenic approach can bring down the cost for iPSC-based cell therapy compared to the autologous approach and will also obviate the need for approval of individual patient-derived products by regulatory authorities [
85]. On the other hand, if the transplanted cells elicit an immune response, the patients will have to be under life-long immunosuppression. Discontinuation of the immunosuppression in these patients will lead to rejection and clearance of the transplanted cells from their organs. Strategies to enable allogenic iPSC-derived therapeutic cells to evade T cell- and natural killer cell-mediated immune responses simultaneously as mentioned in the previous section would favor the use of allogenic iPSC-based cell therapy [
90,
91].
This entry is adapted from the peer-reviewed paper 10.3390/cells8050403