 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Olga Vinogradova | -- | 3192 | 2022-04-25 15:55:27 | | | |
| 2 | Jessie Wu | Meta information modification | 3192 | 2022-04-26 04:00:36 | | | | |
| 3 | Jessie Wu | Meta information modification | 3192 | 2022-04-26 04:03:07 | | |
Video Upload Options
A third of both pro- and eukaryotic proteomes consist of membrane proteins. Housed in a milieu of hydrophobic molecules, they serve as crucial contacts of communication between the cytoplasm and non-cytosolic environments, making them essential pharmaceutical targets. While membrane proteins are notoriously difficult to investigate at any level, high-resolution structures of these targets only became feasible at the very end of the twentieth century. It was not until robust technological developments in the fields of X-ray crystallography, NMR spectroscopy and cryo-EM, that the scientific community at large, finally gained access to an ever-increasing number of atomic resolution structures, and began to rationalize how membrane proteins accommodate their function. As if the lack of structural information wasn’t enough to hamper progress, a higher level of complexity arose from the modern understanding of “one structure—one function” paradigm, a primitive simplification useful at the dawn of the scientific era, that has promptly lost credence to the complex maneuvers of membrane proteins.
2. Nuclear Magnetic Resonance
-
High magnetic sensitivity: the 19F isotope, in contrast to proton (1H), the gold standard, has a high relative sensitivity of ~83% and a 100% natural abundance (note that relative sensitivity for detection in NMR experiments at a constant number of nuclei is roughly proportional to the cube of their gyromagnetic ratios [5], (γF19/γH1)3 = 0.943 = 0.83).
-
The lack of endogenous fluorine and thereby the absence of a background signal.
-
Sensitivity to local environment: 19F exhibits large chemical shifts dispersion (CSD) which spans 2000 ppm in comparison to a meager 13 ppm for 1H; although chemical shifts, arising solely from local van der Waals electrostatic and solvent interactions, typically vary between 2.5 (CF3) and 20 ppms (mono-fluoro-aromatics) [6], they may still be enough to characterize motional and structural properties of IMPs in different environments such as lipid vesicles, detergents or organic solvents.
-
1D spectroscopy, used to avoid unfavorable relaxation associated with multidimensional NMR methods, is usually sufficient for the separation of the peaks in 19F spectrum and works even for potentially dynamic states that are characterized by broad lines. Thus, different states can be resolved, and their corresponding population quantified.
-
Lastly, the lack of protein deuteration significantly improves the ease and efficacy of sample preparations.
3. Cryo-Electron Microscopy
4. Serial X-ray Crystallography
References
- Puthenveetil, R.; Vinogradova, O. Solution NMR: A powerful tool for structural and functional studies of membrane proteins in reconstituted environments. J. Biol. Chem. 2019, 294, 15914–15931.
- Pervushin, K.; Riek, R.; Wider, G.; Wüthrich, K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl. Acad. Sci. USA 1997, 94, 12366–12371.
- Schutz, S.; Sprangers, R. Methyl TROSY spectroscopy: A versatile NMR approach to study challenging biological systems. Prog. Nucl. Magn. Reson. Spectrosc. 2020, 116, 56–84.
- Mooney, E.F.; Winson, P.H. Fluorine-19 Nuclear Magnetic Resonance Spectroscopy. Annu. Rep. NMR Spectrosc. 1968, 1, 243–311.
- Canet, D. NMR: Concepts and Methods; Wiley: New York, NY, USA, 1991.
- Picard, L.P.; Prosser, R.S. Advances in the study of GPCRs by (19)F NMR. Curr. Opin. Struct. Biol. 2021, 69, 169–176.
- Hilger, D.; Masureel, M.; Kobilka, B.K. Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol. 2018, 25, 4–12.
- Huang, S.K.; Pandey, A.; Tran, D.P.; Villanueva, N.L.; Kitao, A.; Sunahara, R.K.; Sljoka, A.; Prosser, R.P. Delineating the conformational landscape of the adenosine A2A receptor during G protein coupling. Cell 2021, 184, 1884–1894.e14.
- Sljoka, A. Probing Allosteric Mechanism with Long-Range Rigidity Transmission Across Protein Networks. Methods Mol. Biol. 2021, 2253, 61–75.
- Di Pietrantonio, C.; Pandey, A.; Gould, J.; Hasabins, A.; Prosser, R.S. Understanding Protein Function Through an Ensemble Description: Characterization of Functional States by (19)F NMR. Methods Enzymol. 2019, 615, 103–130.
- Huang, Y.; Wang, X.; Lv, G.; Razavi, A.M.; Huysmans, G.H.M.; Weinstein, H.; Bracken, C.; Eliezer, D.; Boudker, O. Use of paramagnetic (19)F NMR to monitor domain movement in a glutamate transporter homolog. Nat. Chem. Biol. 2020, 16, 1006–1012.
- Meiboom, S.; Gill, D. Modified SpinEcho Method for Measuring Nuclear Relaxation Times. Rev. Sci. Instrum. 1958, 28, 688–691.
- Zhuravleva, A.; Korzhnev, D.M. Protein folding by NMR. Prog. Nucl. Magn. Reson. Spectrosc. 2017, 100, 52–77.
- Manglik, A.; Kim, T.H.; Masureel, M.; Altenbach, C.; Yang, Z.; Hilger, D.; Lerch, M.T.; Kobilka, T.S.; Thian, F.S.; Hubbell, W.L.; et al. Structural Insights into the Dynamic Process. of beta2-Adrenergic Receptor Signaling. Cell 2015, 161, 1101–1111.
- Boeszoermenyi, A.; Chhabra, S.; Dubey, A.; Radeva, D.L.; Burdzhiev, N.T.; Chanev, C.D.; Petrov, O.I.; Gelev, V.M.; Zhang, M.; Anklin, C.; et al. Aromatic (19)F-(13)C TROSY: A background-free approach to probe biomolecular structure, function, and dynamics. Nat. Methods 2019, 16, 333–340.
- Cellitti, S.E.; Jones, D.H.; Lagpacan, L.; Hao, X.; Zhang, Q.; Hu, H.; Brittain, S.M.; Brinker, A.; Caldwell, J.; Bursulaya, B.; et al. In vivo incorporation of unnatural amino acids to probe structure, dynamics, and ligand binding in a large protein by nuclear magnetic resonance spectroscopy. J. Am. Chem. Soc. 2008, 130, 9268–9281.
- Didenko, T.; Liu, J.J.; Horst, R.; Stevens, R.C.; Wüthrich, K. Fluorine-19 NMR of integral membrane proteins illustrated with studies of GPCRs. Curr. Opin. Struct. Biol. 2013, 23, 740–747.
- Yang, F.; Yu, X.; Liu, C.; Qu, C.-X.; Gong, Z.; Liu, H.-D.; Li, F.-H.; Wang, H.-M.; He, D.-F.; Yi, F.; et al. Phospho-selective mechanisms of arrestin conformations and functions revealed by unnatural amino acid incorporation and (19)F-NMR. Nat. Commun. 2015, 6, 8202.
- Kuhlbrandt, W. Biochemistry. The resolution revolution. Science 2014, 343, 1443–1444.
- Hite, R.K.; MacKinnon, R. Structural Titration of Slo2.2, a Na+-Dependent K+ Channel. Cell 2017, 168, 390–399.e11.
- Liao, M.; Cao, E.; Julius, D.; Cheng, Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 2013, 504, 107–112.
- Cao, E.; Liao, M.; Cheng, Y.; Julius, Y. TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 2013, 504, 113–118.
- Paulsen, C.E.; Armache, J.P.; Gao, Y.; Cheng, Y.; Julius, D. Structure of the TRPA1 ion channel suggests regulatory mechanisms. Nature 2015, 525, 552.
- Jin, P.; Bulkley, D.; Guo, Y.; Zhang, W.; Guo, Z.; Huynh, W.; Wu, S.; Meltzer, S.; Cheng, T.; Jan, L.Y.; et al. Electron. cryo-microscopy structure of the mechanotransduction channel NOMPC. Nature 2017, 547, 118–122.
- Hirschi, M.; Herzik, M.A., Jr.; Wie, J.; Suo, Y.; Borschel, W.F.; Ren, D.; Lander, G.C.; Lee, S.-Y. Cryo-electron microscopy structure of the lysosomal calcium-permeable channel TRPML3. Nature 2017, 550, 411–414.
- Chen, Q.; She, J.; Zeng, W.; Guo, J.; Xu, H.; Bai, X.C.; Jiang, Y. Structure of mammalian endolysosomal TRPML1 channel in nanodiscs. Nature 2017, 550, 415–418.
- Zhang, K.; Julius, D.; Cheng, Y. Structural snapshots of TRPV1 reveal mechanism of polymodal functionality. Cell 2021, 184, 5138–5150.e12.
- Steinberg, X.; Kasimova, M.A.; Cabezas-Bratesco, D.; Galpin, J.D.; Ladron-de-Guevara, E.; Villa, F.; Carnevale, V.; Islas, L.; Ahern, C.A.; Brauchi, S.E. Conformational dynamics in TRPV1 channels reported by an encoded coumarin amino acid. eLife 2017, 6, e28626.
- Chen, I.; Pant, S.; Wu, Q.; Cater, R.J.; Sobti, M.; Vandenberg, R.J.; Stewart, A.G.; Tajkhorshid, E.; Font, J.; Ryan, R.M. Glutamate transporters have a chloride channel with two hydrophobic gates. Nature 2021, 591, 327–331.
- Cater, R.J.; Ryan, R.M.; Vandenberg, R.J. The Split Personality of Glutamate Transporters: A Chloride Channel and a Transporter. Neurochem. Res. 2016, 41, 593–599.
- Arkhipova, V.; Guskov, A.; Slotboom, D.J. Structural ensemble of a glutamate transporter homologue in lipid nanodisc environment. Nat. Commun. 2020, 11, 998.
- Ge, J.; Elferich, J.; Dehghani-Ghahnaviyeh, S.; Zhao, Z.; Meadows, M.; von Gersdorff, H.; Tajkhorshid, E.; Gouaux, E. Molecular mechanism of prestin electromotive signal amplification. Cell 2021, 184, 4669–4679.e13.
- Bavi, N.; Clark, M.D.; Contreras, G.F.; Shen, R.; Reddy, B.G.; Milewski, W.; Perozo, E. The conformational cycle of prestin underlies outer-hair cell electromotility. Nature 2021, 600, 553–558.
- Butan, C.; Song, Q.; Bai, J.-P.; Tan, W.J.Y.; Navaratnam, D.; Santos-Sacchi, J. Single particle cryo-EM structure of the outer hair cell motor protein prestin. Nat. Commun. 2022, 13, 290.
- Santos-Sacchi, J.; Shen, W.; Zheng, J.; Dallos, P. Effects of membrane potential and tension on prestin, the outer hair cell lateral membrane motor protein. J. Physiol. 2001, 531, 661–666.
- Schumacher, S.; Deddebm, D.; Nunez, R.V.; Matoba, K.; Takagi, J.; Biertümpfel, C.; Mizuno, N. Structural insights into integrin alpha5beta1 opening by fibronectin ligand. Sci. Adv. 2021, 7, eabe9716.
- Klebl, D.P.; White, H.D.; Sobott, F.; Muench, S.P. On-grid and in-flow mixing for time-resolved cryo-EM. Acta Crystallogr. D Struct. Biol. 2021, 77, 1233–1240.
- Kaledhonkar, S.; Fu, Z.; White, H.; Frank, J. Time-Resolved Cryo-electron Microscopy Using a Microfluidic Chip. Methods Mol. Biol. 2018, 1764, 59–71.
- Maeots, M.E.; Lee, B.; Nans, A.; Jeong, S.-G.; Esfahani, M.M.N.; Ding, S.; Smith, D.J.; Lee, C.-S.; Lee, S.S.; Peter, M.; et al. Modular microfluidics enables kinetic insight from time-resolved cryo-EM. Nat. Commun. 2020, 11, 3465.
- Dandey, V.P.; Budell, W.C.; Wei, H.; Bobe, D.; Maruthi, K.; Kopylov, M.; Eng, E.T.; Kahn, P.A.; Hinshaw, J.E.; Kundu, N.; et al. Time-resolved cryo-EM using Spotiton. Nat. Methods 2020, 17, 897–900.
- Shaikh, T.R.; Barnard, D.; Meng, X.; Wagenknecht, T. Implementation of a flash-photolysis system for time-resolved cryo-electron microscopy. J. Struct. Biol. 2009, 165, 184–189.
- Yoder, N.; Jalali-Yazdi, F.; Noreng, S.; Houser, A.; Baconguis, I.; Gouaux, E. Light-coupled cryo-plunger for time-resolved cryo-EM. J. Struct. Biol. 2020, 212, 107624.
- Weinert, T.; Skopintsev, P.; James, D.; Dworkowski, F.; Panepucci, E.; Kekilli, D.; Furrer, A.; Brünle, S.; Mous, S.; Ozerov, D.; et al. Proton uptake mechanism in bacteriorhodopsin captured by serial synchrotron crystallography. Science 2019, 365, 61–65.
- Martin-Garcia, J.M. Protein Dynamics and Time Resolved Protein Crystallography at Synchrotron Radiation Sources: Past, Present and Future. Crystals 2021, 11, 521.
- Wickstrand, C.; Nogly, P.; Nango, E.; Iwata, S.; Standfuss, J.; Neutze, R. Bacteriorhodopsin: Structural Insights Revealed Using X-ray Lasers and Synchrotron Radiation. Annu. Rev. Biochem. 2019, 88, 59–83.
- Baxter, R.H.; Ponomarenko, N.; Srajer, V.; Pahl, R.; Moffat, K.; Norris, J.R. Time-resolved crystallographic studies of light-induced structural changes in the photosynthetic reaction center. Proc. Natl. Acad. Sci. USA 2004, 101, 5982–5987.
- Wohri, A.B.; Katona, G.; Johansson, L.C.; Fritz, E.; Malmerberg, E.; Andersson, M.; Vincent, J.; Eklund, M.; Cammarata, M.; Wulff, M.; et al. Light-induced structural changes in a photosynthetic reaction center caught by Laue diffraction. Science 2010, 328, 630–633.
- Neutze, R.; Wouts, R.; van der Spoel, D.; Weckert, E.; Hajdu, J. Potential for biomolecular imaging with femtosecond X-ray pulses. Nature 2000, 406, 752–757.
- Aquila, A.; Hunter, M.S.; Doak, R.B.; Kirian, R.A.; Fromme, P.; White, T.A.; Andreasson, J.; Arnlund, D.; Bajt, S.; Barends, T.R.M.; et al. Time-resolved protein nanocrystallography using an X-ray free-electron laser. Opt. Express 2012, 20, 2706–2716.
- Kern, J.; Alonso-Mori, R.; Tran, R.; Hattne, J.; Gildea, R.J.; Echols, N.; Glöckner, C.; Hellmich, J.; Laksmono, H.; Sierra, R.G.; et al. Simultaneous femtosecond X-ray spectroscopy and diffraction of photosystem II at room temperature. Science 2013, 340, 491–495.
- Kupitz, C.; Basu, S.; Grotjohann, I.; Fromme, R.; Zatsepin, N.A.; Rendek, K.N.; Hunter, M.S.; Shoeman, R.L.; White, T.A.; Wang, D.; et al. Serial time-resolved crystallography of photosystem II using a femtosecond X-ray laser. Nature 2014, 513, 261–265.
- Kern, J.; Tran, R.; Alonso-Mori, R.; Koroidov, S.; Echols, N.; Hattne, J.; Ibrahim, M.; Gul, S.; Laksmono, H.; Sierra, R.G.; et al. Taking snapshots of photosynthetic water oxidation using femtosecond X-ray diffraction and spectroscopy. Nat. Commun. 2014, 5, 4371.
- Suga, M.; Akita, F.; Hirata, K.; Ueno, G.; Murakami, H.; Nakajima, Y.; Shimizu, T.; Yamashita, K.; Yamamoto, M.; Ago, H.; et al. Native structure of photosystem II at 1.95 A resolution viewed by femtosecond X-ray pulses. Nature 2015, 517, 99–103.
- Young, I.D.; Ibrahim, M.; Chatterjee, R.; Gul, S.; Fuller, F.; Koroidov, S.; Brewster, A.S.; Tran, R.; Alonso-Mori, R.; Kroll, T.; et al. Structure of photosystem II and substrate binding at room temperature. Nature 2016, 540, 453–457.
- Sauter, N.K.; Echols, N.; Adams, P.D.; Zwart, P.H.; Kern, J.; Brewster, A.S.; Koroidov, S.; Alonso-Mori, R.; Zouni, A.; Messinger, J.; et al. No observable conformational changes in PSII. Nature 2016, 533, E1–E2.
- Suga, M.; Akita, F.; Sugahara, M.; Kubo, M.; Nakajima, Y.; Nakane, T.; Yamashita, K.; Umena, Y.; Nakabayashi, M.; Yamane, T.; et al. Light-induced structural changes and the site of O = O bond formation in PSII caught by XFEL. Nature 2017, 543, 131–135.
- Kern, J.; Chatterjee, R.; Young, I.D.; Fuller, F.D.; Lassalle, L.; Ibrahim, M.; Gul, S.; Fransson, T.; Brewster, A.S.; Alonso-Mori, R.; et al. Structures of the intermediates of Kok’s photosynthetic water oxidation clock. Nature 2018, 563, 421–425.
- Suga, M.; Akita, F.; Yamashita, K.; Nakajima, Y.; Ueno, G.; Li, H.; Yamane, T.; Hirata, K.; Umena, Y.; Yonekura, S.; et al. An oxyl/oxo mechanism for oxygen-oxygen coupling in PSII revealed by an X-ray free-electron laser. Science 2019, 366, 334–338.
- Ibrahim, M.; Fransson, T.; Chatterjee, R.; Cheah, M.H.; Hussein, R.; Lassalle, L.; Sutherlin, K.D.; Young, I.D.; Fuller, F.D.; Gul, S.; et al. Untangling the sequence of events during the S2 --> S3 transition in photosystem II and implications for the water oxidation mechanism. Proc. Natl. Acad. Sci. USA 2020, 117, 12624–12635.
- Hussein, R.; Ibrahim, M.; Bhowmick, A.; Simon, P.S.; Chatterjee, R.; Lassalle, L.; Doyle, M.; Bogacz, I.; Kim, I.-S.; Cheah, M.H.; et al. Structural dynamics in the water and proton channels of photosystem II during the S2 to S3 transition. Nat. Commun. 2021, 12, 6531.
- Bao, H.; Burnap, R.L. Photoactivation: The Light-Driven Assembly of the Water Oxidation Complex. of Photosystem II. Front. Plant Sci. 2016, 7, 578.
- Kok, B.; Forbush, B.; McGloin, M. Cooperation of charges in photosynthetic O2 evolution-I. A linear four step mechanism. Photochem. Photobiol. 1970, 11, 457–475.
- Dods, R.; Båth, P.; Morozov, D.; Gagnér, V.A.; Arnlund, D.; Luk, H.L.; Kübel, J.; Maj, M.; Vallejos, A.; Wickstrand, C.; et al. Ultrafast structural changes within a photosynthetic reaction centre. Nature 2021, 589, 310–314.
- Lanyi, J.K. Bacteriorhodopsin. Annu. Rev. Physiol. 2004, 66, 665–688.
- Nango, E.; Royant, A.; Kubo, M.; Nakane, T.; Wickstrand, C.; Kimura, T.; Tanaka, T.; Tono, K.; Song, C.; Tanaka, R.; et al. A three-dimensional movie of structural changes in bacteriorhodopsin. Science 2016, 354, 1552–1557.
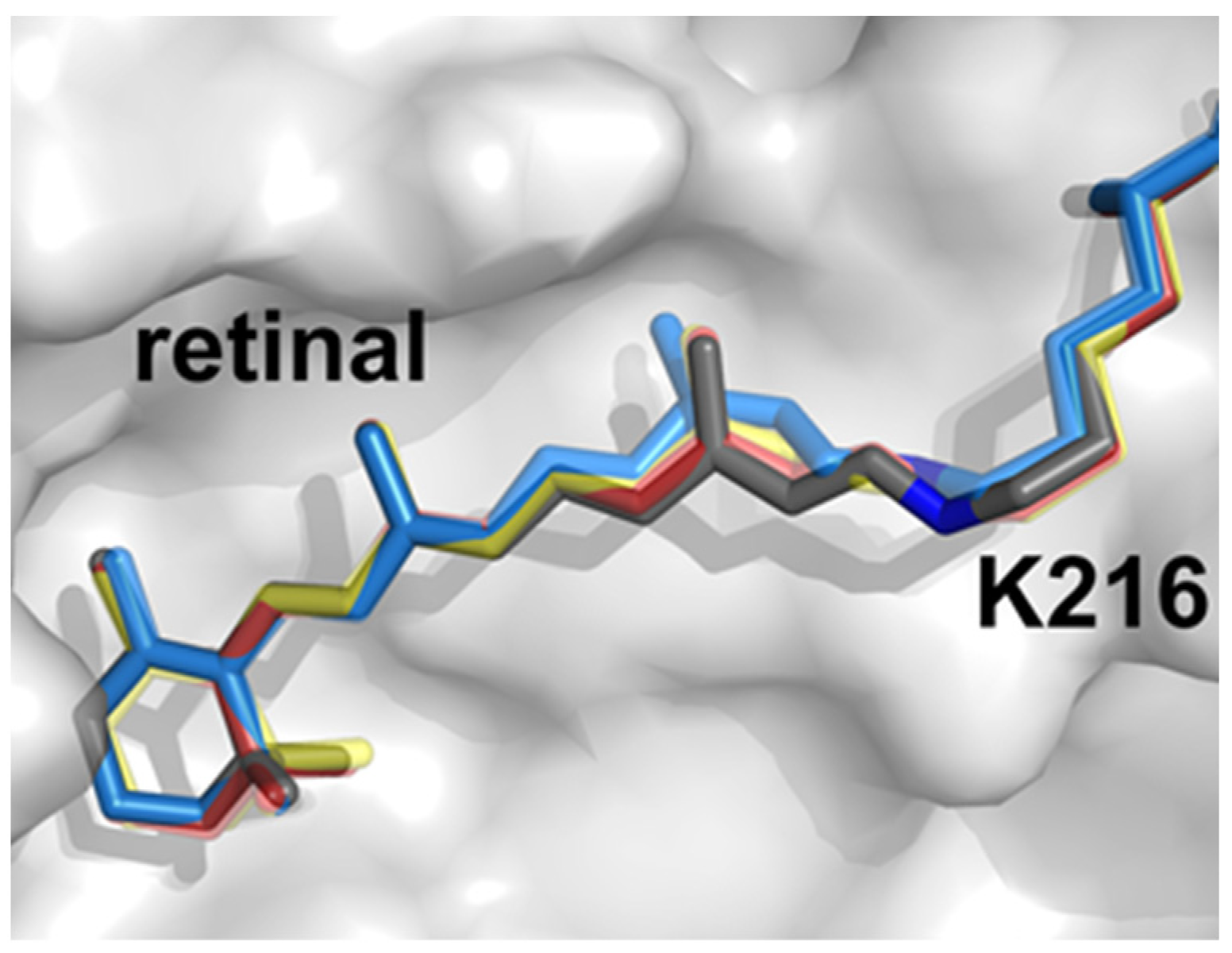
- Nogly, P.; Weinert, T.; James, D.; Carbajo, S.; Ozerov, D.; Furrer, A.; Gashi, D.; Borin, V.; Skopintsev, P.; Jaeger, K.; et al. Retinal isomerization in bacteriorhodopsin captured by a femtosecond X-ray laser. Science 2018, 361, eaat0094.
- Kovacs, G.N.; Colletier, J.P.; Grünbein, M.L.; Yang, Y.; Stensitzki, T.; Batyuk, A.; Carbajo, S.; Doak, R.B.; Ehrenberg, D.; Foucar, L.; et al. Three-dimensional view of ultrafast dynamics in photoexcited bacteriorhodopsin. Nat. Commun. 2019, 10, 3177.
- Skopintsev, P.; Ehrenberg, D.; Weinert, T.; James, D.; Kar, R.K.; Johnson, P.J.M.; Ozerov, D.; Furrer, A.; Martiel, I.; Dworkowski, F.; et al. Femtosecond-to-millisecond structural changes in a light-driven sodium pump. Nature 2020, 583, 314–318.
- Yun, J.H.; Li, X.; Yue, J.; Park, J.-H.; Jin, Z.; Li, C.; Hu, H.; Shim, Y.; Pandey, S.; Carbajo, S.; et al. Early-stage dynamics of chloride ion-pumping rhodopsin revealed by a femtosecond X-ray laser. Proc. Natl. Acad. Sci. USA 2021, 118, e2020486118.
- Oda, K.; Nomura, T.; Nakane, T.; Yamashita, K.; Inoue, K.; Ito, S.; Vierock, J.; Hirata, K.; Maturana, A.D.; Katayama, K.; et al. Time-resolved serial femtosecond crystallography reveals early structural changes in channelrhodopsin. eLife 2021, 10, e62389.
- Shimada, A.; Kubo, M.; Baba, S.; Yamashita, K.; Hirata, K.; Ueno, G.; Nomura, T.; Kimura, T.; Shinzawa-Itoh, K.; Baba, J.; et al. A nanosecond time-resolved XFEL analysis of structural changes associated with CO release from cytochrome c oxidase. Sci. Adv. 2017, 3, e1603042.
- Ishigami, I.; Lewis-Ballester, A.; Echelmeier, A.; Brehm, G.; Zatsepin, N.A.; Grant, T.D.; Coe, J.D.; Lisova, S.; Nelson, G.; Zhang, S.; et al. Snapshot of an oxygen intermediate in the catalytic reaction of cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 2019, 116, 3572–3577.
- Gisriel, C.; Coe, J.; Letrun, R.; Yefanov, O.M.; Luna-Chavez, C.; Stander, N.E.; Lisova, S.; Mariani, V.; Kuhn, M.; Aplin, S.; et al. Membrane protein megahertz crystallography at the European XFEL. Nat. Commun. 2019, 10, 5021.


